

Translating slow-binding inhibition kinetics into cellular and in vivo effects
- PMID: 25894085
- PMCID: PMC4536915
- DOI: 10.1038/nchembio.1796
Translating slow-binding inhibition kinetics into cellular and in vivo effects
Abstract
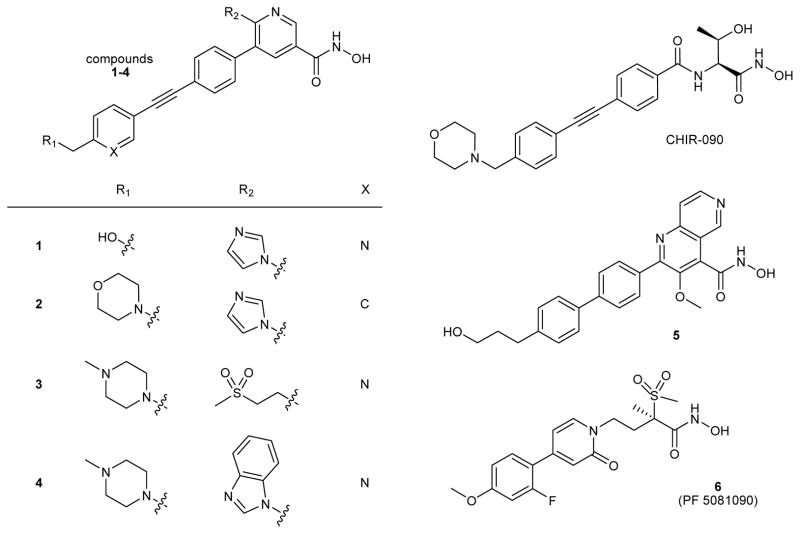
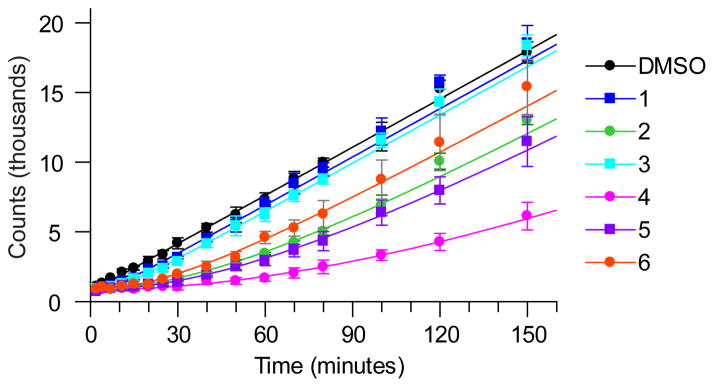
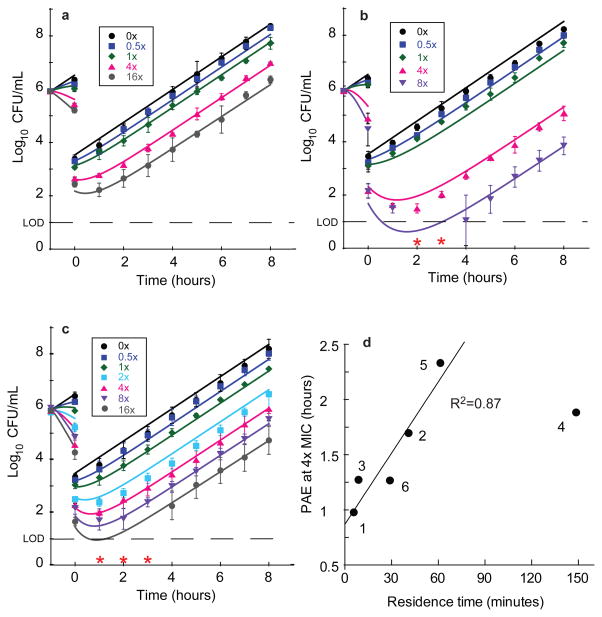
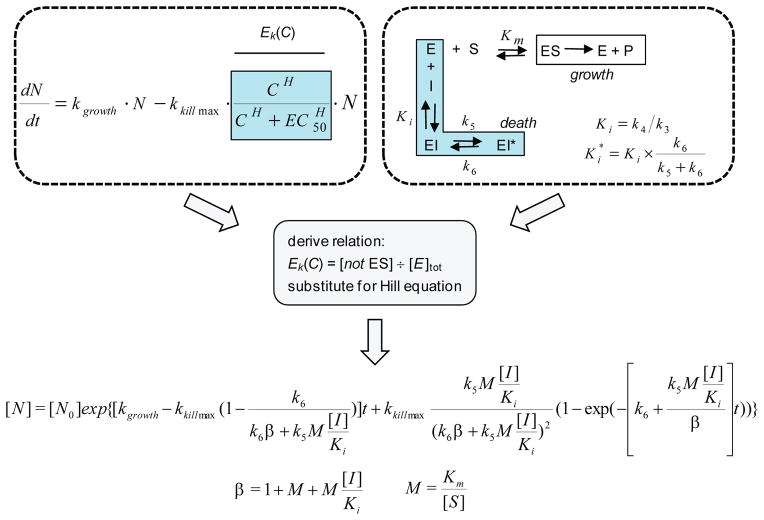
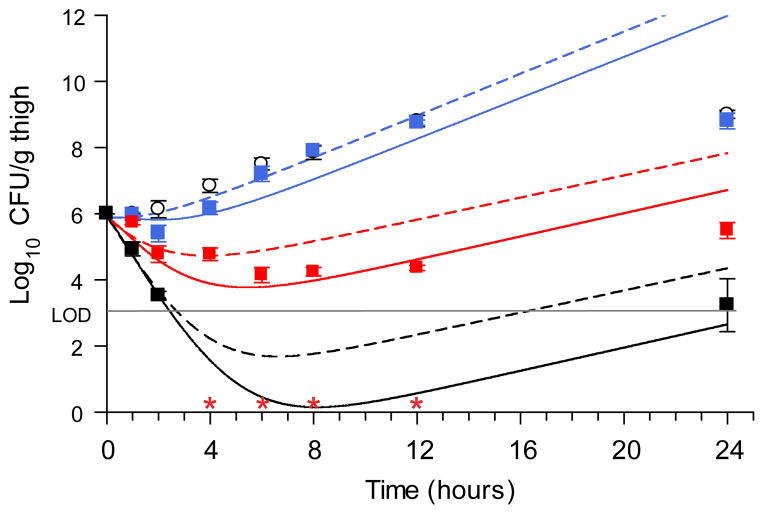
Many drug candidates fail in clinical trials owing to a lack of efficacy from limited target engagement or an insufficient therapeutic index. Minimizing off-target effects while retaining the desired pharmacodynamic (PD) response can be achieved by reduced exposure for drugs that display kinetic selectivity in which the drug-target complex has a longer half-life than off-target-drug complexes. However, though slow-binding inhibition kinetics are a key feature of many marketed drugs, prospective tools that integrate drug-target residence time into predictions of drug efficacy are lacking, hindering the integration of drug-target kinetics into the drug discovery cascade. Here we describe a mechanistic PD model that includes drug-target kinetic parameters, including the on- and off-rates for the formation and breakdown of the drug-target complex. We demonstrate the utility of this model by using it to predict dose response curves for inhibitors of the LpxC enzyme from Pseudomonas aeruginosa in an animal model of infection.
Conflict of interest statement
All authors except E.K.H.A, F.D., S.G.W., and P.J.T. were employees of AstraZeneca during the conduct of this research.
Figures
Comment in
-
Pharmacodynamics: Which trails are your drugs taking?Nat Chem Biol. 2015 Jun;11(6):382-3. doi: 10.1038/nchembio.1795. Epub 2015 Apr 20. Nat Chem Biol. 2015. PMID: 25894084 No abstract available.
References
-
- Swinney DC. Biochemical Mechanisms of New Molecular Entities (NMEs) Approved by United States FDA During 2001–2004: Mechanisms Leading to Optimal Efficacy and Safety. Curr Top Med Chem. 2006;6:461–478. - PubMed
-
- Swinney DC. The role of binding kinetics in therapeutically useful drug action. Curr Opin Drug Discov Devel. 2009;12:31–39. - PubMed
-
- Arrowsmith J. Trial watch: Phase II failures: 2008–2010. Nat Rev Drug Discov. 2011;10:328. - PubMed
-
- Cook D, et al. Lessons learned from the fate of AstraZeneca’s drug pipeline: a five-dimensional framework. Nat Rev Drug Discov. 2014;13:419–431. - PubMed
-
- Morgan P, et al. Can the flow of medicines be improved? Fundamental pharmacokinetic and pharmacological principles toward improving Phase II survival. Drug Discov Today. 2012;17:419–24. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
