

High-resolution crystal structures of protein helices reconciled with three-centered hydrogen bonds and multipole electrostatics
- PMID: 25894612
- PMCID: PMC4403875
- DOI: 10.1371/journal.pone.0123146
High-resolution crystal structures of protein helices reconciled with three-centered hydrogen bonds and multipole electrostatics
Abstract
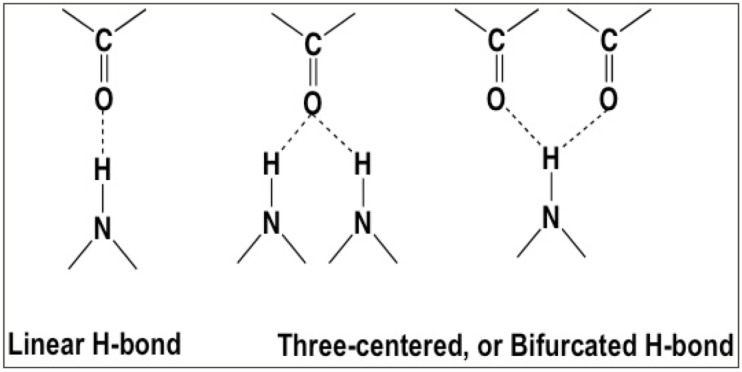
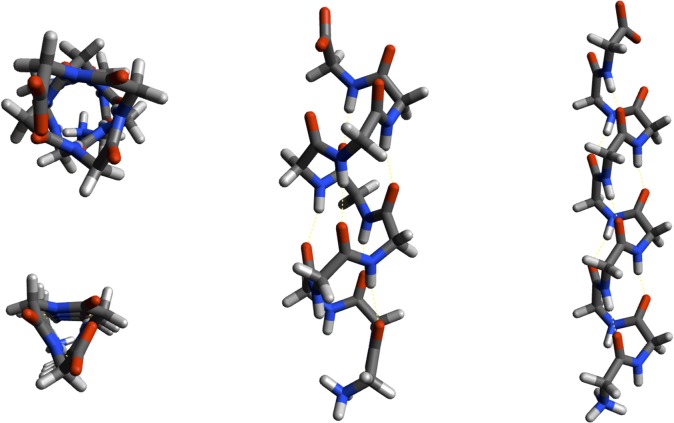
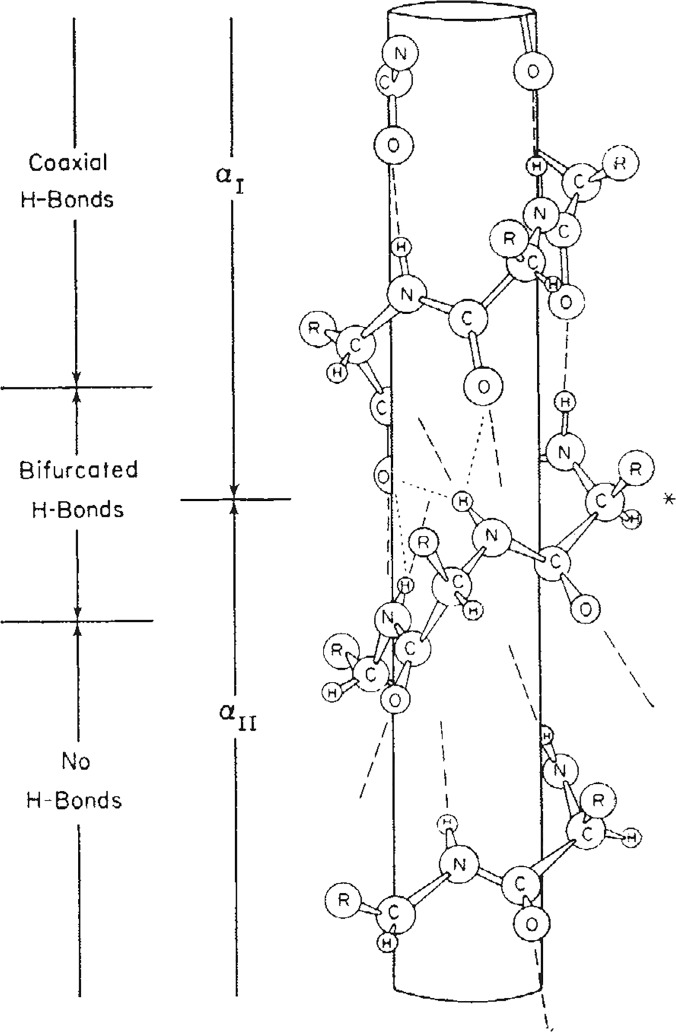
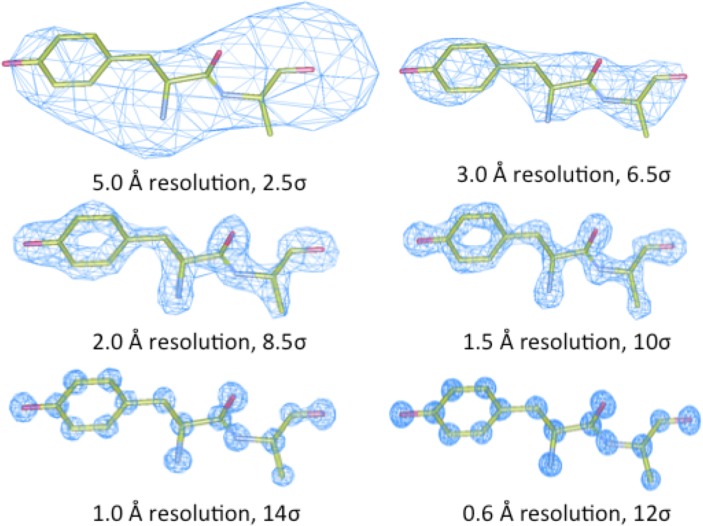
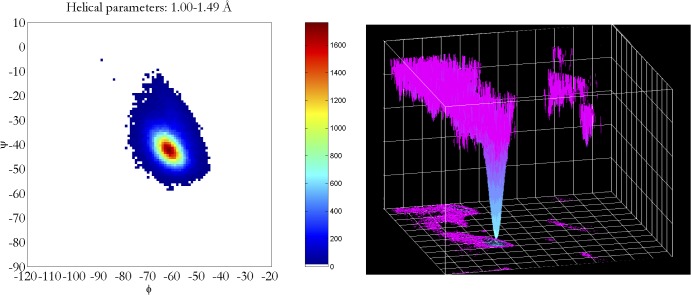
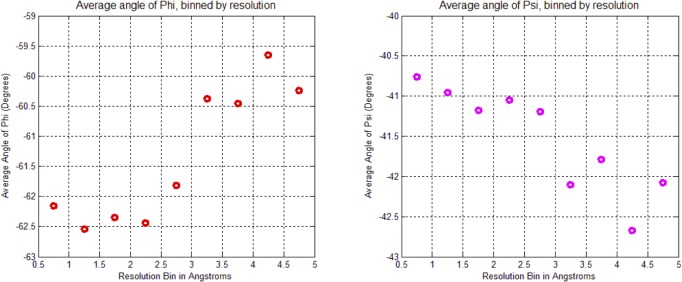
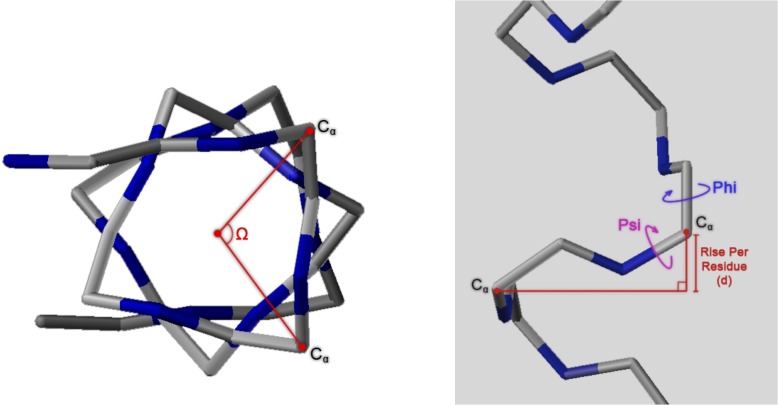
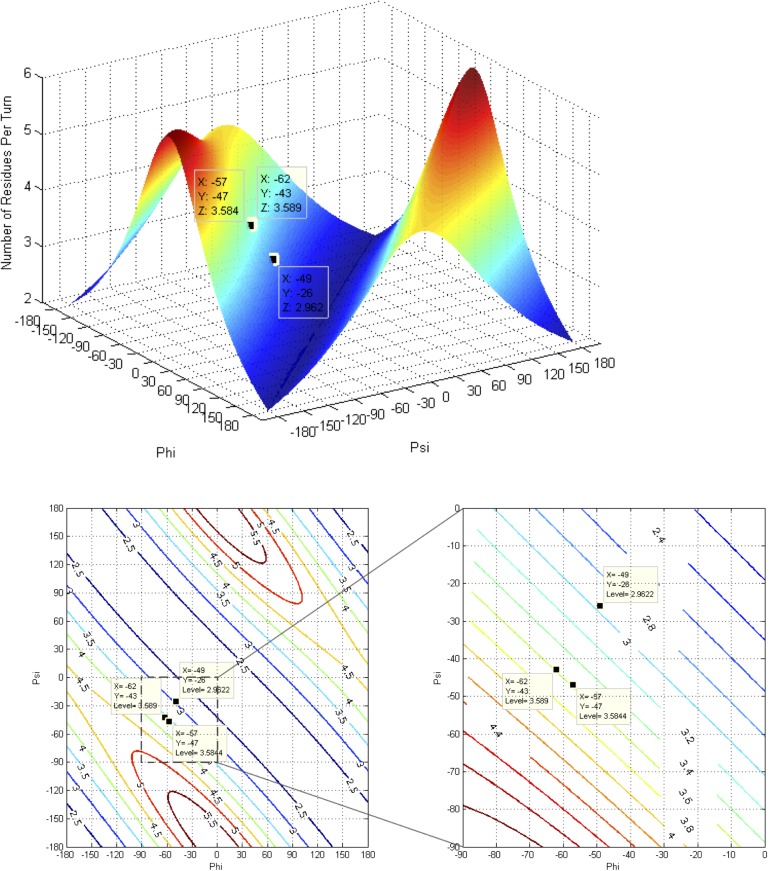
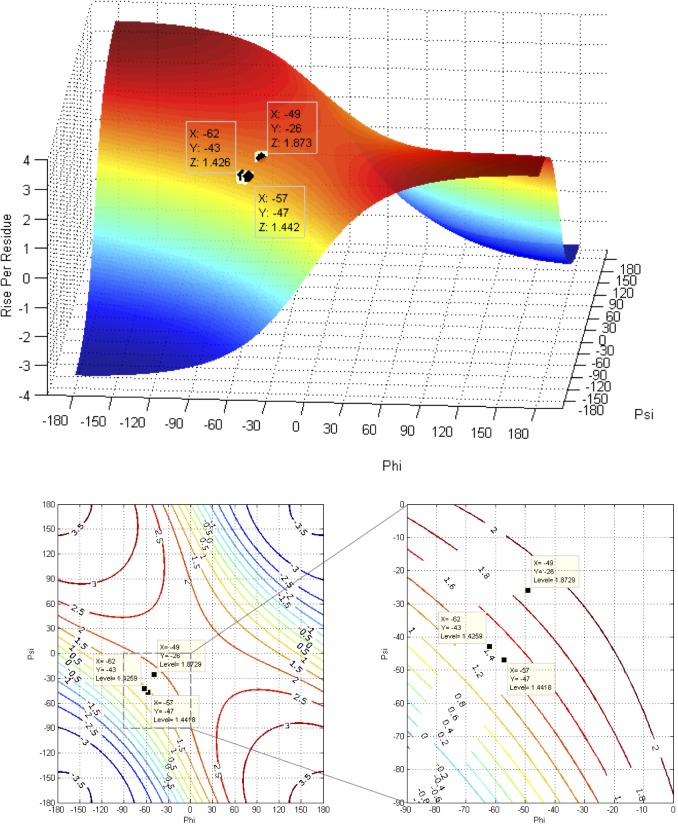
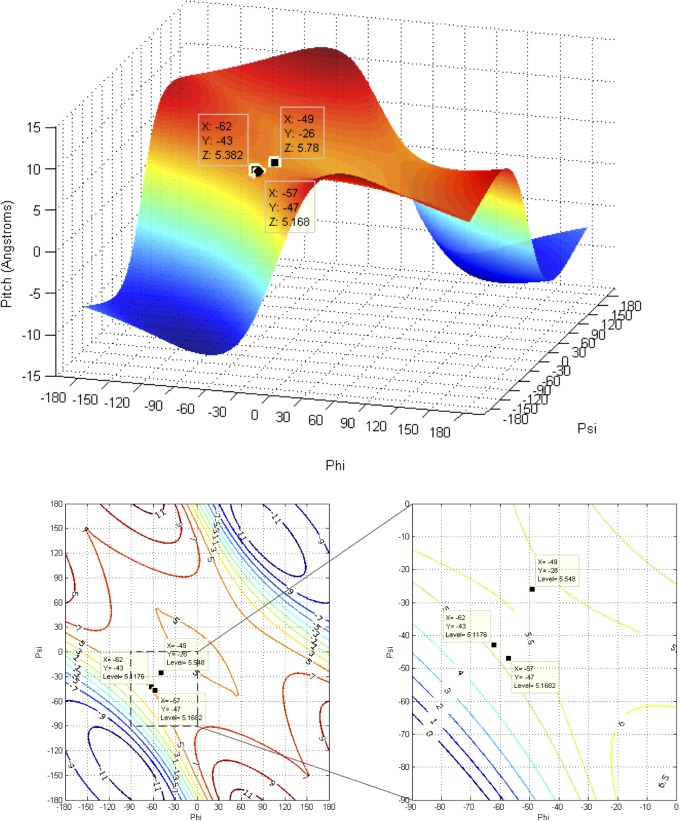
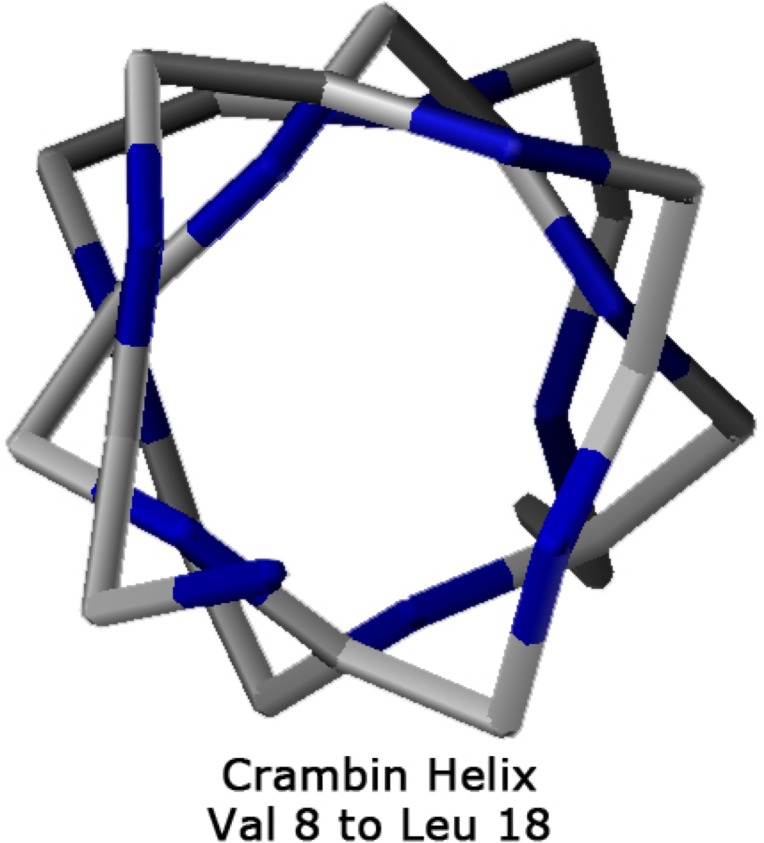
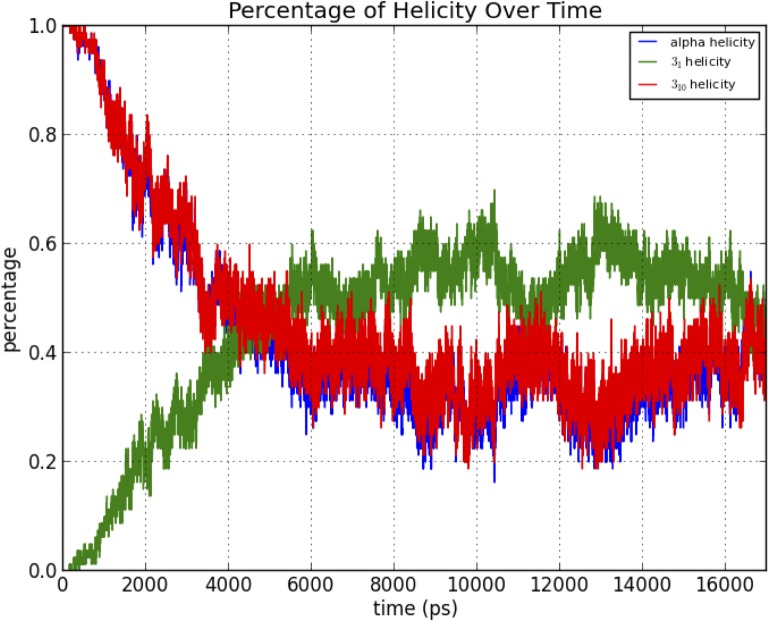
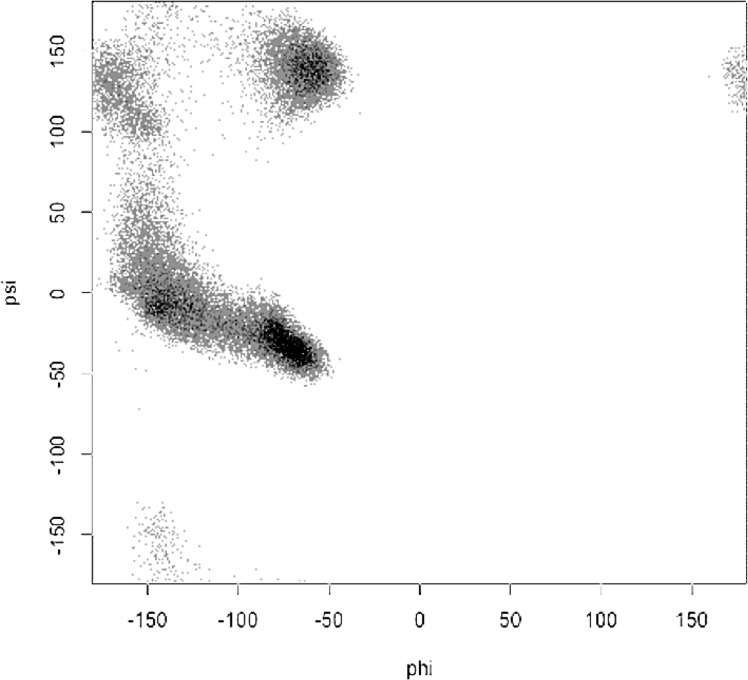
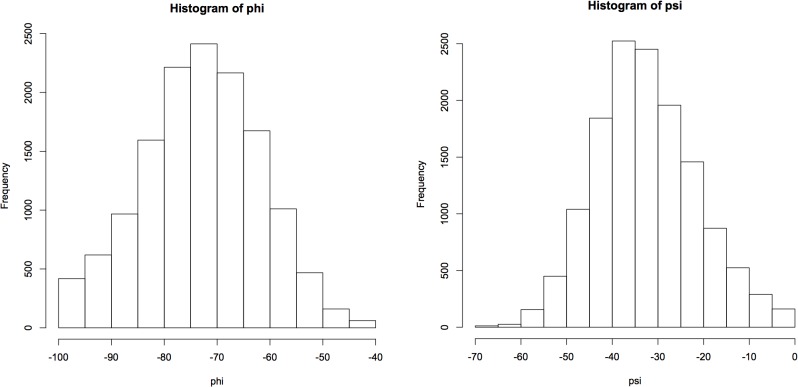
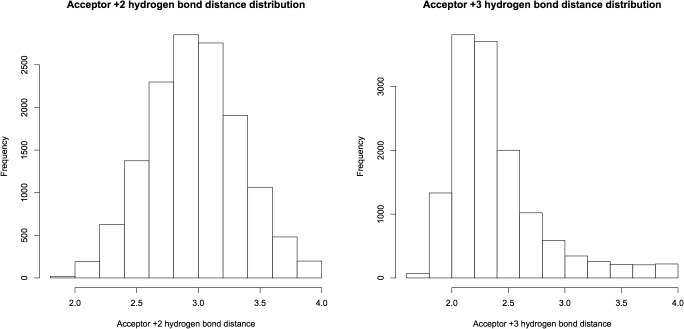
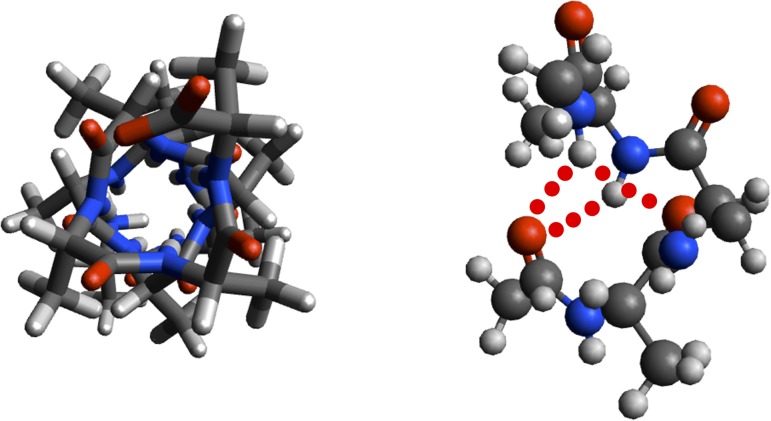
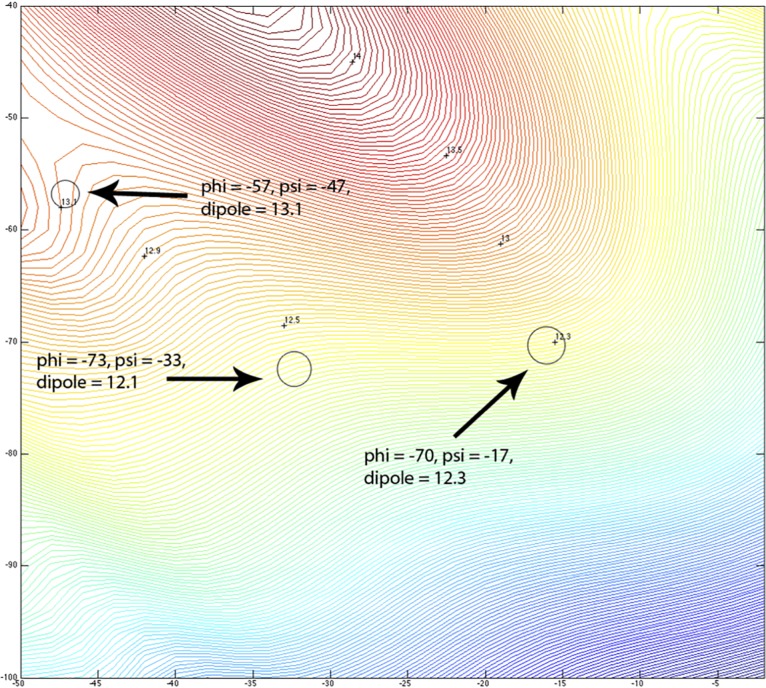
Theoretical and experimental evidence for non-linear hydrogen bonds in protein helices is ubiquitous. In particular, amide three-centered hydrogen bonds are common features of helices in high-resolution crystal structures of proteins. These high-resolution structures (1.0 to 1.5 Å nominal crystallographic resolution) position backbone atoms without significant bias from modeling constraints and identify Φ = -62°, ψ = -43 as the consensus backbone torsional angles of protein helices. These torsional angles preserve the atomic positions of α-β carbons of the classic Pauling α-helix while allowing the amide carbonyls to form bifurcated hydrogen bonds as first suggested by Némethy et al. in 1967. Molecular dynamics simulations of a capped 12-residue oligoalanine in water with AMOEBA (Atomic Multipole Optimized Energetics for Biomolecular Applications), a second-generation force field that includes multipole electrostatics and polarizability, reproduces the experimentally observed high-resolution helical conformation and correctly reorients the amide-bond carbonyls into bifurcated hydrogen bonds. This simple modification of backbone torsional angles reconciles experimental and theoretical views to provide a unified view of amide three-centered hydrogen bonds as crucial components of protein helices. The reason why they have been overlooked by structural biologists depends on the small crankshaft-like changes in orientation of the amide bond that allows maintenance of the overall helical parameters (helix pitch (p) and residues per turn (n)). The Pauling 3.6(13) α-helix fits the high-resolution experimental data with the minor exception of the amide-carbonyl electron density, but the previously associated backbone torsional angles (Φ, Ψ) needed slight modification to be reconciled with three-atom centered H-bonds and multipole electrostatics. Thus, a new standard helix, the 3.6(13/10)-, Némethy- or N-helix, is proposed. Due to the use of constraints from monopole force fields and assumed secondary structures used in low-resolution refinement of electron density of proteins, such structures in the PDB often show linear hydrogen bonding.
Conflict of interest statement
Figures
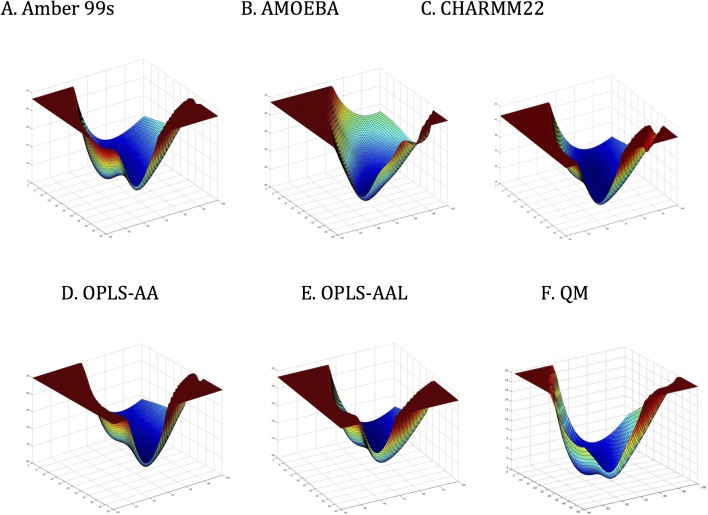
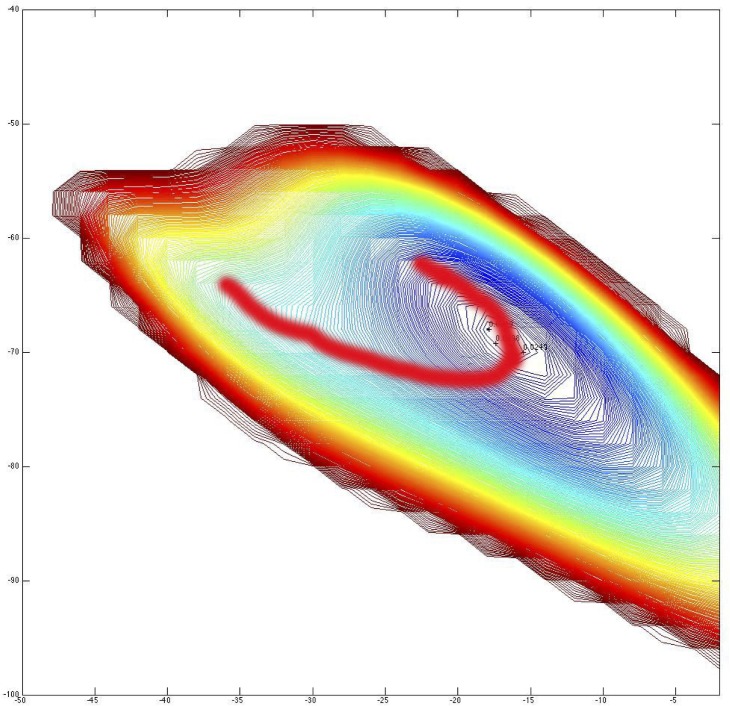
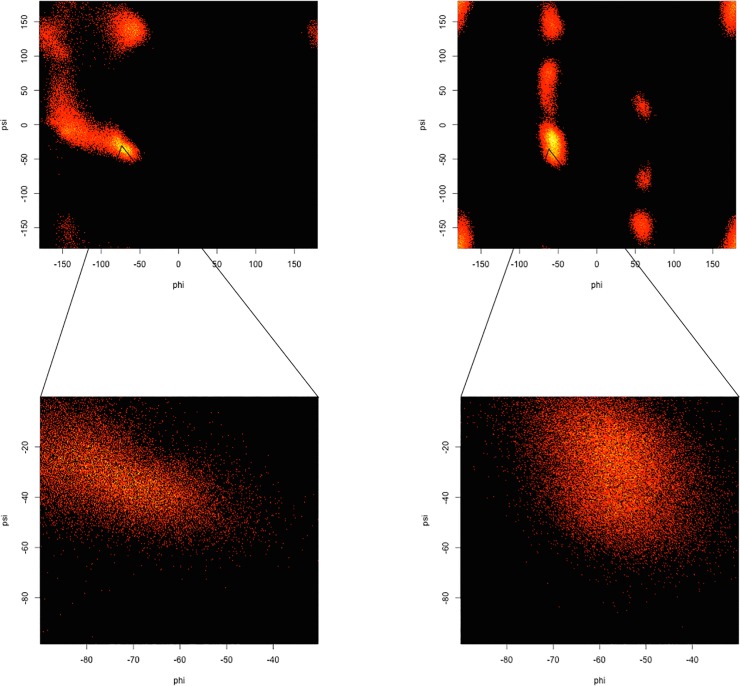
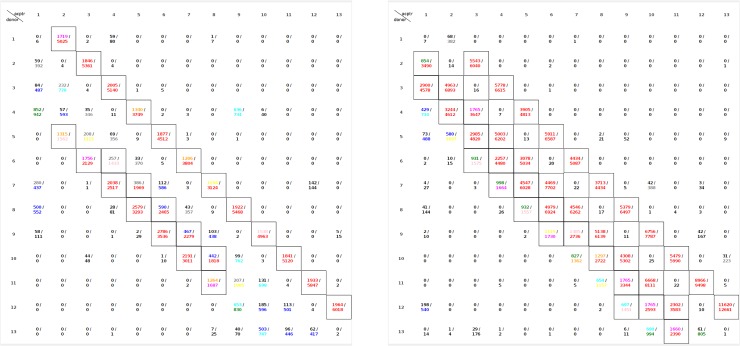
Similar articles
-
3(10)-helices in proteins are parahelices.Proteins. 2006 Aug 15;64(3):691-9. doi: 10.1002/prot.21026. Proteins. 2006. PMID: 16783793
-
Influence of the environment in the conformation of alpha-helices studied by protein database search and molecular dynamics simulations.Biophys J. 2002 Jun;82(6):3207-13. doi: 10.1016/S0006-3495(02)75663-4. Biophys J. 2002. PMID: 12023245 Free PMC article.
-
Peptide bonds revisited.IUCrJ. 2025 May 1;12(Pt 3):307-321. doi: 10.1107/S2052252525002106. IUCrJ. 2025. PMID: 40162645 Free PMC article.
-
Roles of electrostatic interaction in proteins.Q Rev Biophys. 1996 Feb;29(1):1-90. doi: 10.1017/s0033583500005746. Q Rev Biophys. 1996. PMID: 8783394 Review.
-
Biomolecular force fields: where have we been, where are we now, where do we need to go and how do we get there?J Comput Aided Mol Des. 2019 Feb;33(2):133-203. doi: 10.1007/s10822-018-0111-4. Epub 2018 Nov 30. J Comput Aided Mol Des. 2019. PMID: 30506158 Review.
Cited by
-
De novo design of discrete, stable 310-helix peptide assemblies.Nature. 2022 Jul;607(7918):387-392. doi: 10.1038/s41586-022-04868-x. Epub 2022 Jun 22. Nature. 2022. PMID: 35732733
-
n→π* Interactions Are Competitive with Hydrogen Bonds.Org Lett. 2016 Aug 5;18(15):3614-7. doi: 10.1021/acs.orglett.6b01655. Epub 2016 Jul 13. Org Lett. 2016. PMID: 27409515 Free PMC article.
-
Small Changes: Using Assessment to Direct Instructional Practices in Large-Enrollment Biochemistry Courses.CBE Life Sci Educ. 2017 Spring;16(1):ar7. doi: 10.1187/cbe.16-06-0191. CBE Life Sci Educ. 2017. PMID: 28188280 Free PMC article.
-
Comparative functional properties of engineered cationic antimicrobial peptides consisting exclusively of tryptophan and either lysine or arginine.J Med Microbiol. 2016 Jun;65(6):554-565. doi: 10.1099/jmm.0.000258. Epub 2016 Apr 5. J Med Microbiol. 2016. PMID: 27046192 Free PMC article.
-
Salt-bridge networks within globular and disordered proteins: characterizing trends for designable interactions.J Mol Model. 2017 Jul;23(7):206. doi: 10.1007/s00894-017-3376-y. Epub 2017 Jun 19. J Mol Model. 2017. PMID: 28626846
References
-
- Pauling L, Corey RB (1950) Two Hydrogen-Bonded Spiral Configurations of the Polypeptide Chain. J Am Chem Soc: 5349.
-
- (1970) IUPAC-IUB Commission on Biochemical Nomenclature. Abbreviations and symbols for the description of the conformation of polypeptide chains. Tentative rules (1969). Biochemistry 9: 3471–3479. - PubMed
-
- Arnott S, Dover SD (1967) Refinement of bond angles of an alpha-helix. J Mol Biol 30: 209–212. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
