

Building a better fragment library for de novo protein structure prediction
- PMID: 25901595
- PMCID: PMC4406757
- DOI: 10.1371/journal.pone.0123998
Building a better fragment library for de novo protein structure prediction
Abstract
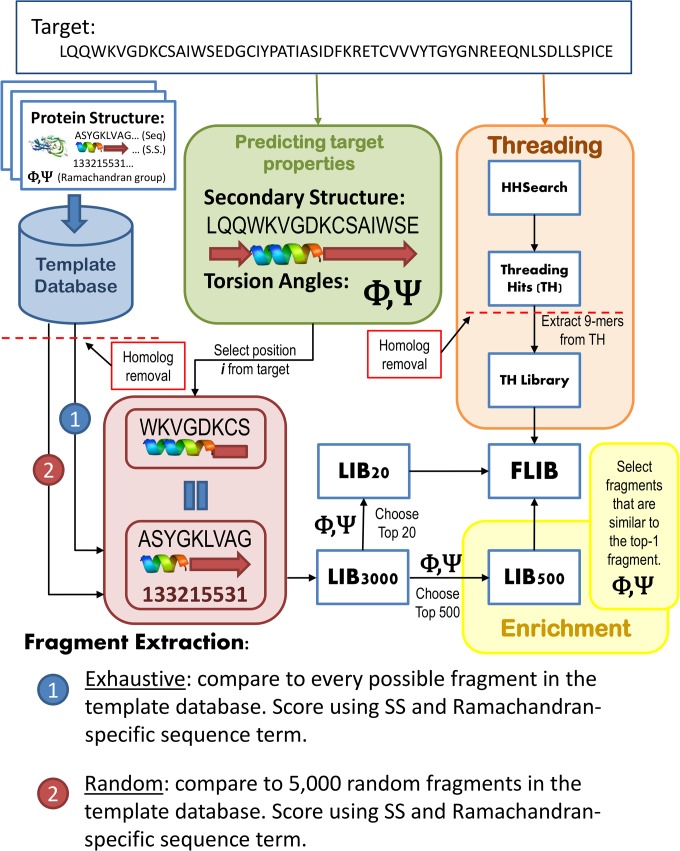
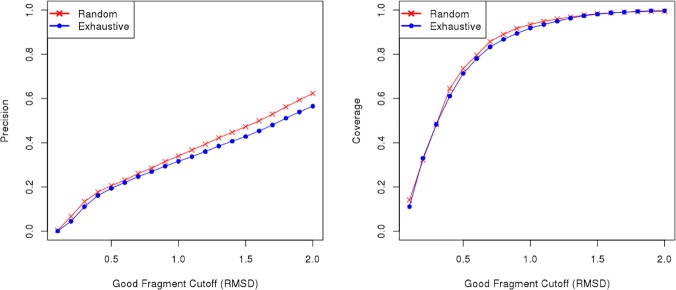
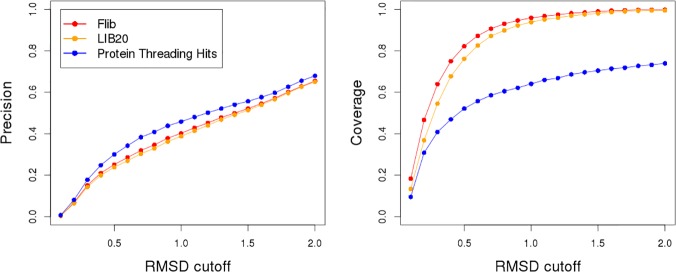
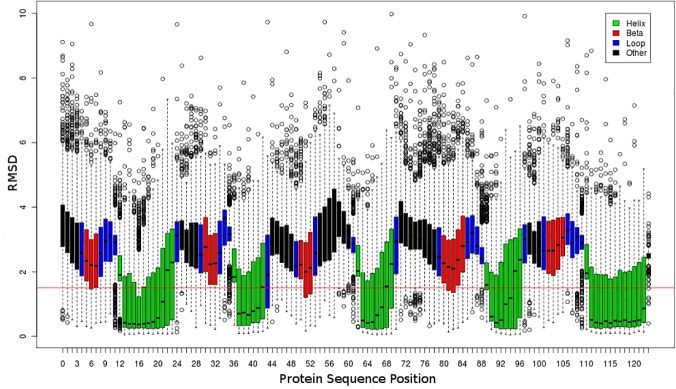
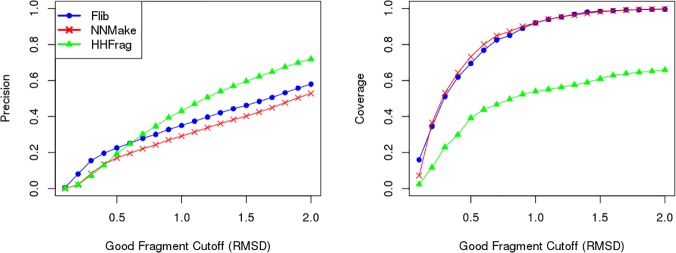
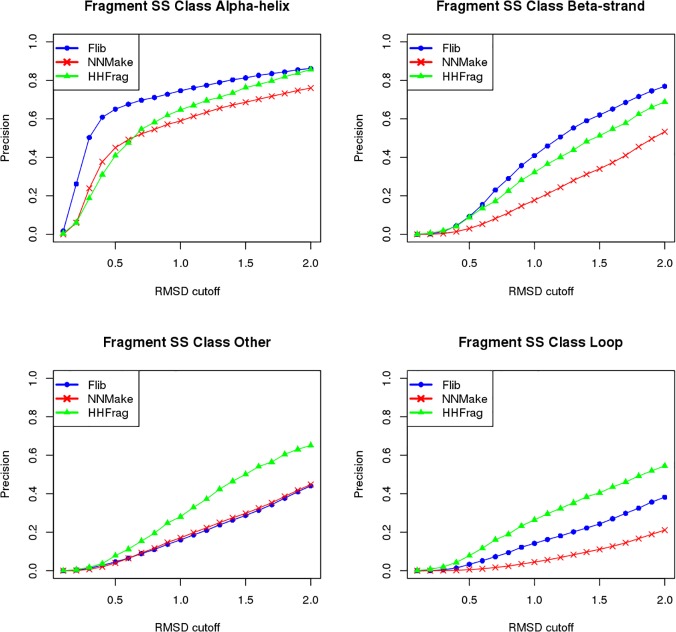
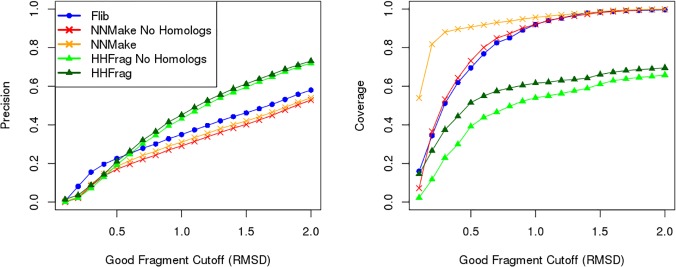
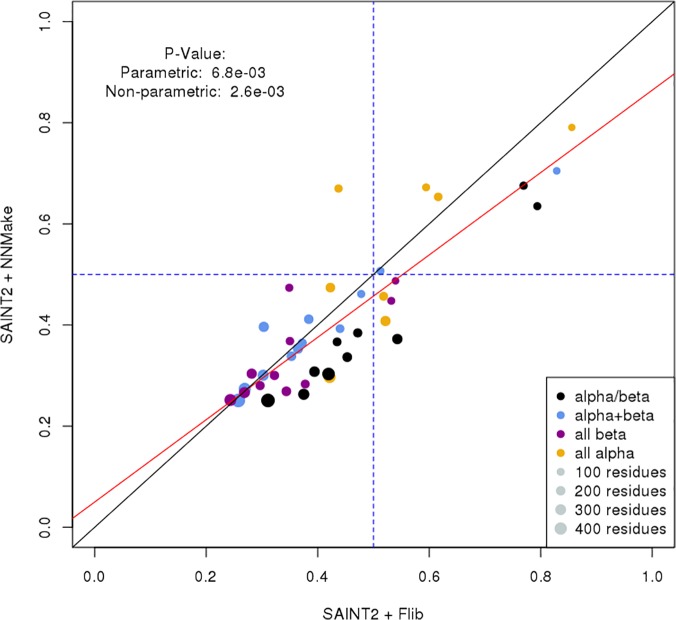
Fragment-based approaches are the current standard for de novo protein structure prediction. These approaches rely on accurate and reliable fragment libraries to generate good structural models. In this work, we describe a novel method for structure fragment library generation and its application in fragment-based de novo protein structure prediction. The importance of correct testing procedures in assessing the quality of fragment libraries is demonstrated. In particular, the exclusion of homologs to the target from the libraries to correctly simulate a de novo protein structure prediction scenario, something which surprisingly is not always done. We demonstrate that fragments presenting different predominant predicted secondary structures should be treated differently during the fragment library generation step and that exhaustive and random search strategies should both be used. This information was used to develop a novel method, Flib. On a validation set of 41 structurally diverse proteins, Flib libraries presents both a higher precision and coverage than two of the state-of-the-art methods, NNMake and HHFrag. Flib also achieves better precision and coverage on the set of 275 protein domains used in the two previous experiments of the the Critical Assessment of Structure Prediction (CASP9 and CASP10). We compared Flib libraries against NNMake libraries in a structure prediction context. Of the 13 cases in which a correct answer was generated, Flib models were more accurate than NNMake models for 10. "Flib is available for download at: http://www.stats.ox.ac.uk/research/proteins/resources".
Conflict of interest statement
Figures
References
-
- Bradley P, Misura KM, Baker D. Toward high-resolution de novo structure prediction for small proteins. Science 309, 1868–71. (2005) - PubMed
-
- Bonneau R, Strauss CE, Rohl CA, Chivian D, Bradley P, Malmstrom L et al. De novo prediction of three-dimensional structures for major protein families. J Mol Biol 322(1):65–78 (2002) - PubMed
-
- Bonneau R, Tsai J, Ruczinski I, Chivian D, Rohl C, Strauss CE et al. Rosetta in CASP4: progress in ab initio protein structure prediction. Proteins Suppl 5:119–26 (2001) - PubMed
Publication types
MeSH terms
Substances
Associated data
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
