

Genome-wide negative feedback drives transgenerational DNA methylation dynamics in Arabidopsis
- PMID: 25902052
- PMCID: PMC4406451
- DOI: 10.1371/journal.pgen.1005154
Genome-wide negative feedback drives transgenerational DNA methylation dynamics in Arabidopsis
Abstract
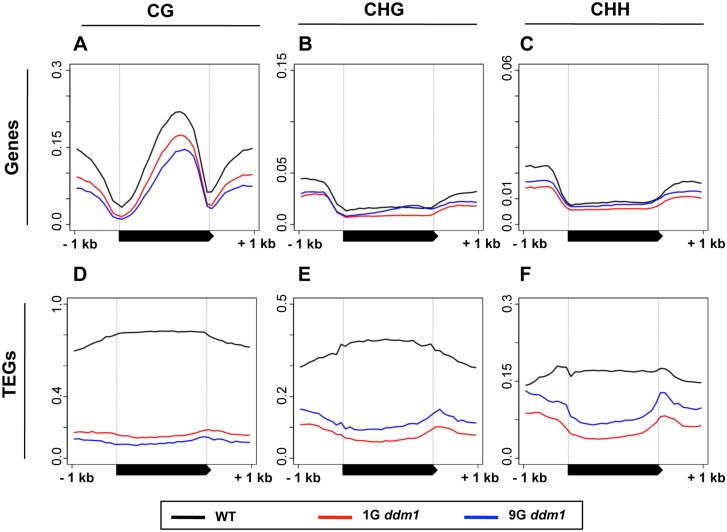
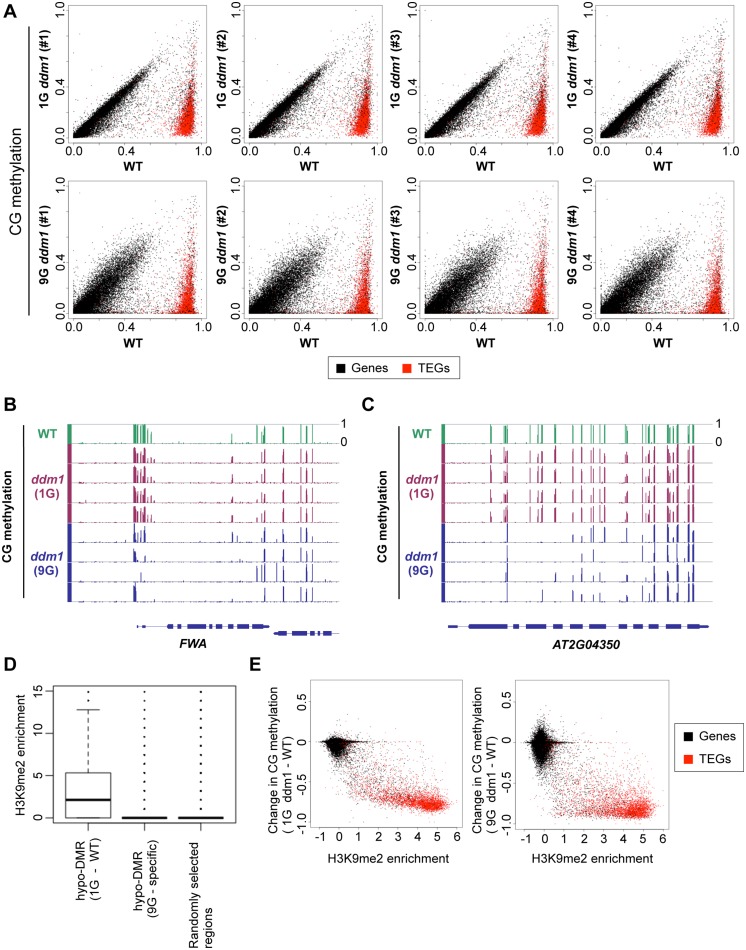
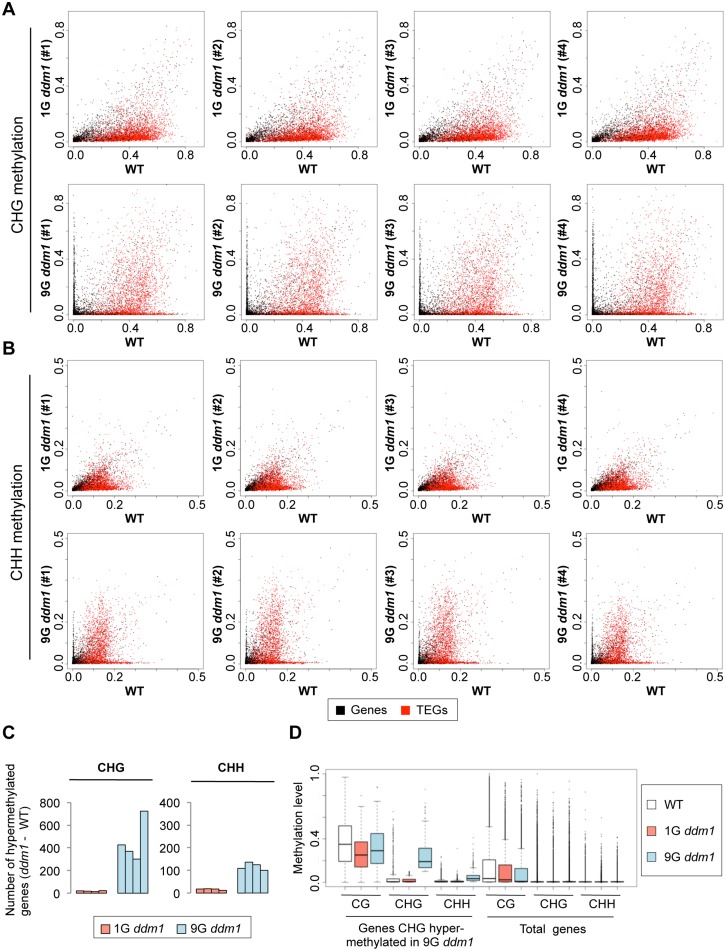
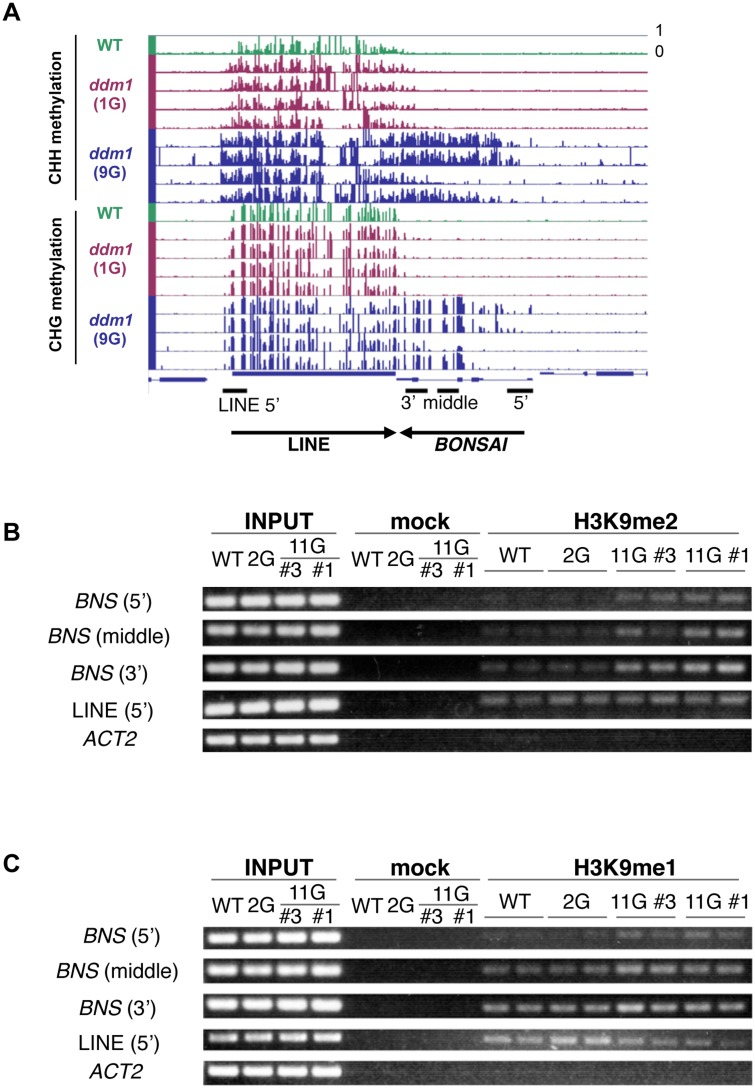
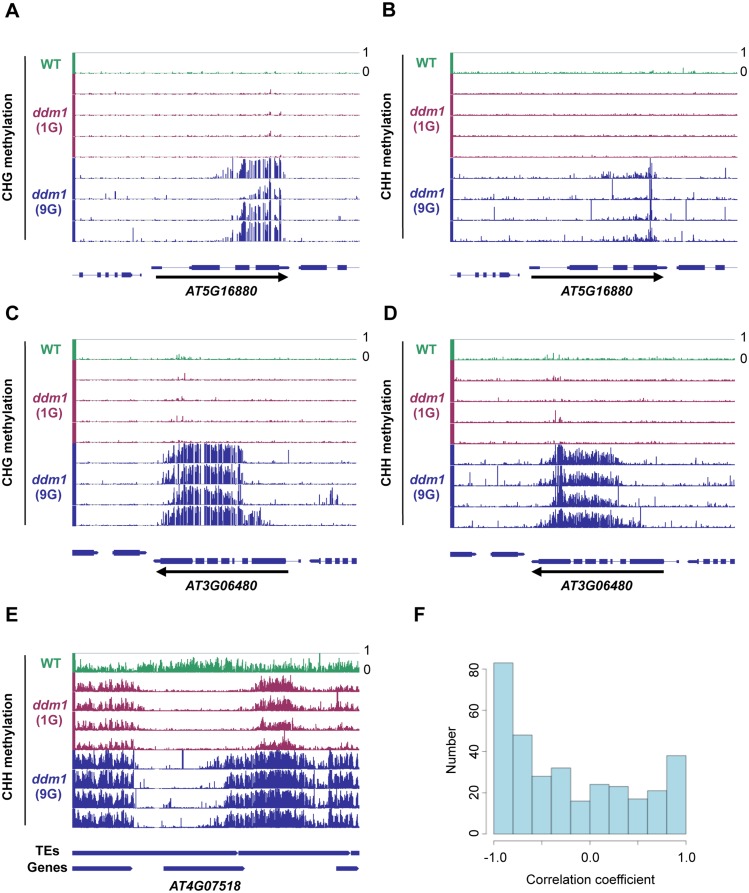
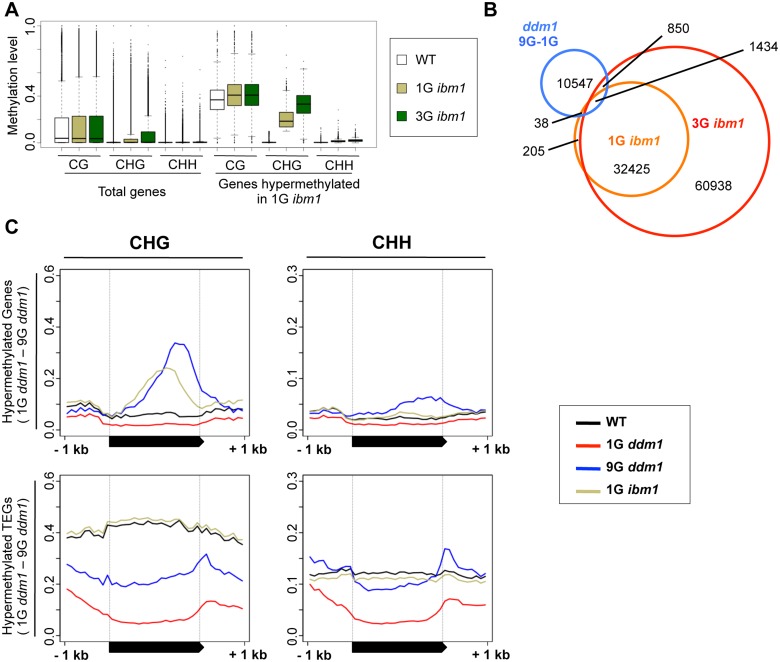
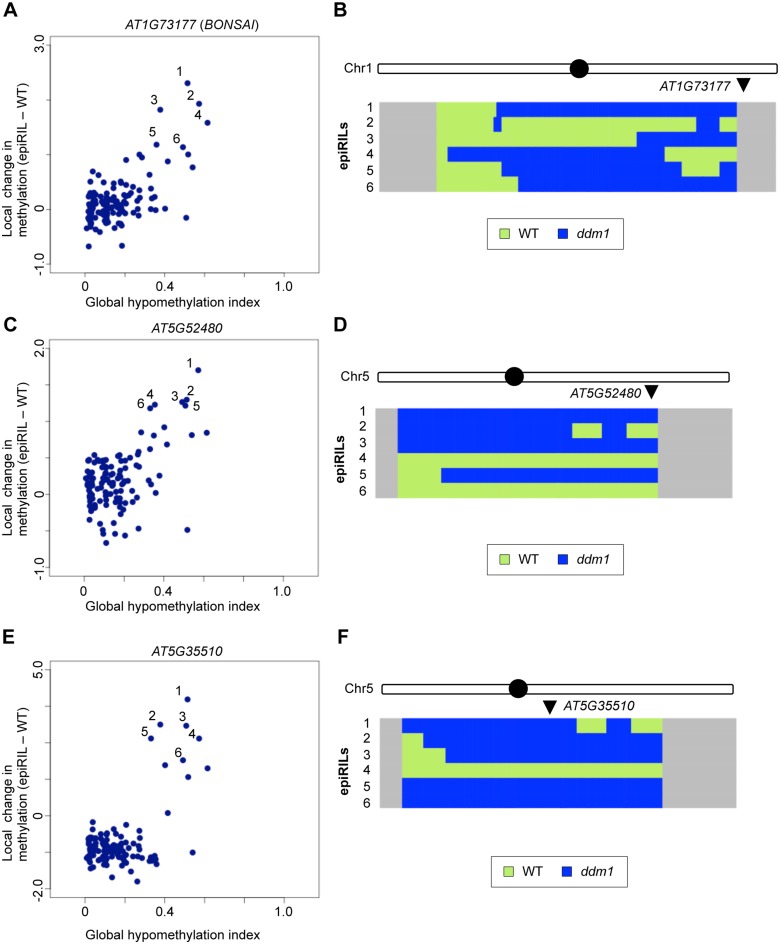
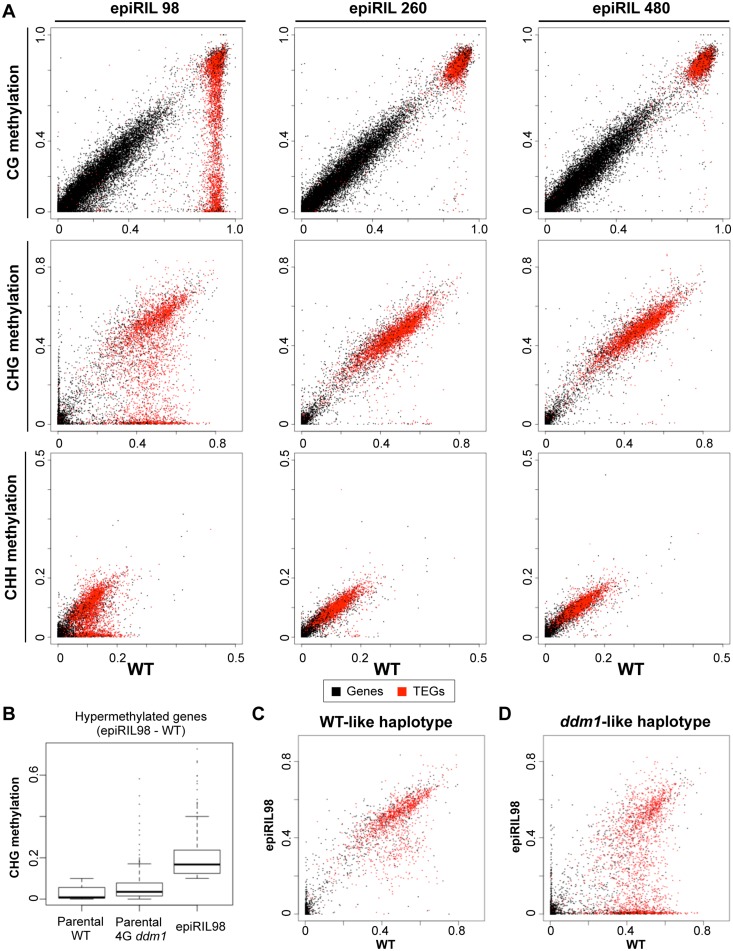
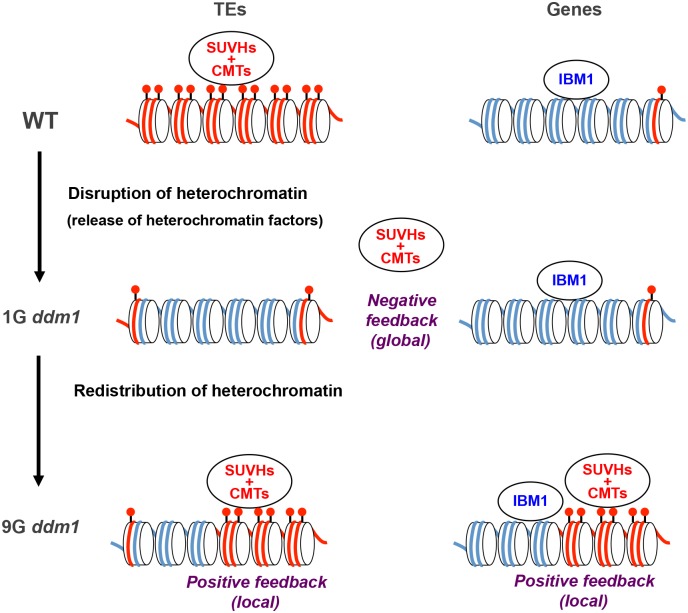
Epigenetic variations of phenotypes, especially those associated with DNA methylation, are often inherited over multiple generations in plants. The active and inactive chromatin states are heritable and can be maintained or even be amplified by positive feedback in a transgenerational manner. However, mechanisms controlling the transgenerational DNA methylation dynamics are largely unknown. As an approach to understand the transgenerational dynamics, we examined long-term effect of impaired DNA methylation in Arabidopsis mutants of the chromatin remodeler gene DDM1 (Decrease in DNA Methylation 1) through whole genome DNA methylation sequencing. The ddm1 mutation induces a drastic decrease in DNA methylation of transposable elements (TEs) and repeats in the initial generation, while also inducing ectopic DNA methylation at hundreds of loci. Unexpectedly, this ectopic methylation can only be seen after repeated self-pollination. The ectopic cytosine methylation is found primarily in the non-CG context and starts from 3' regions within transcription units and spreads upstream. Remarkably, when chromosomes with reduced DNA methylation were introduced from a ddm1 mutant into a DDM1 wild-type background, the ddm1-derived chromosomes also induced analogous de novo accumulation of DNA methylation in trans. These results lead us to propose a model to explain the transgenerational DNA methylation redistribution by genome-wide negative feedback. The global negative feedback, together with local positive feedback, would ensure robust and balanced differentiation of chromatin states within the genome.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
References
-
- Kakutani T (2002) Epi-alleles in plants: inheritance of epigenetic information over generations. Plant Cell Physiol 43: 1106–1111. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
