

Mapping and analysis of Caenorhabditis elegans transcription factor sequence specificities
- PMID: 25905672
- PMCID: PMC4434323
- DOI: 10.7554/eLife.06967
Mapping and analysis of Caenorhabditis elegans transcription factor sequence specificities
Abstract
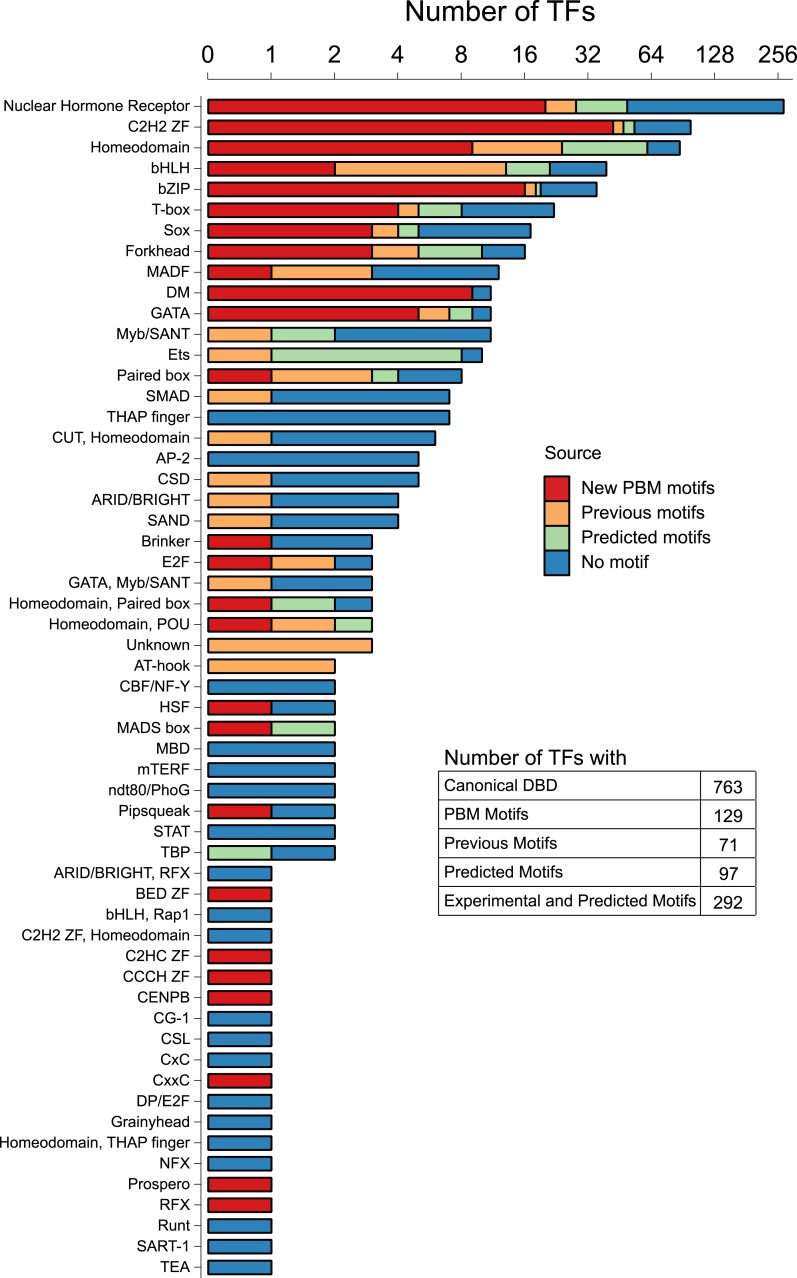
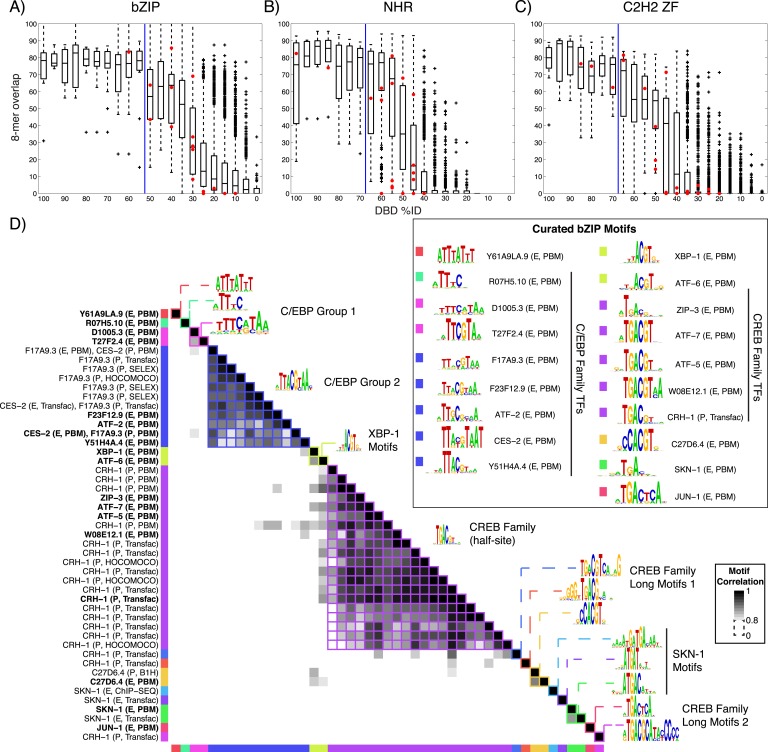
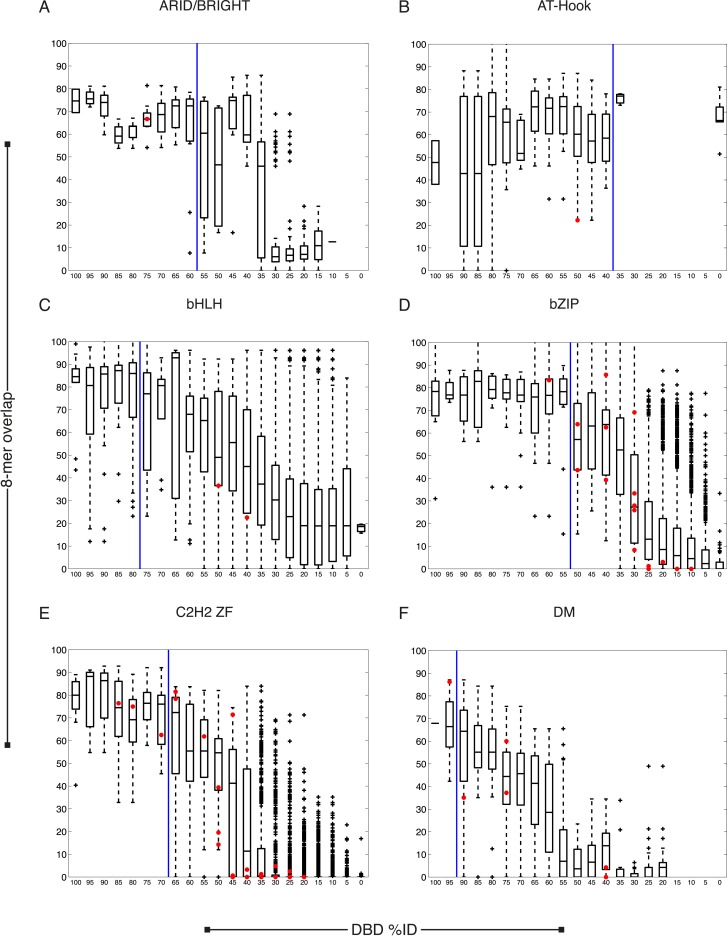
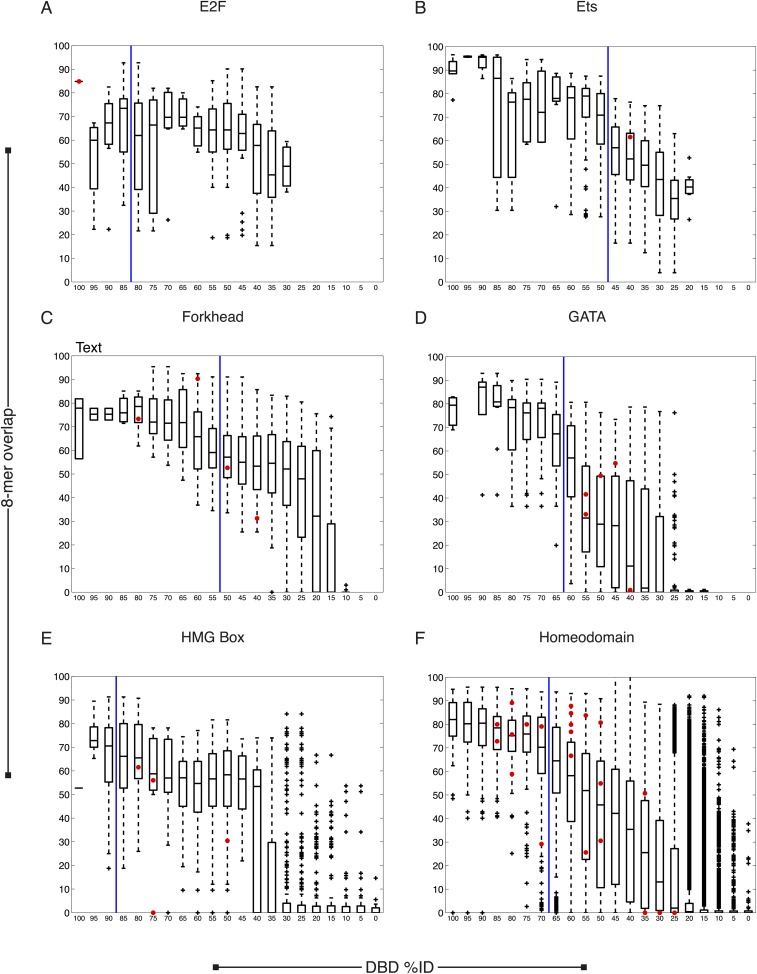
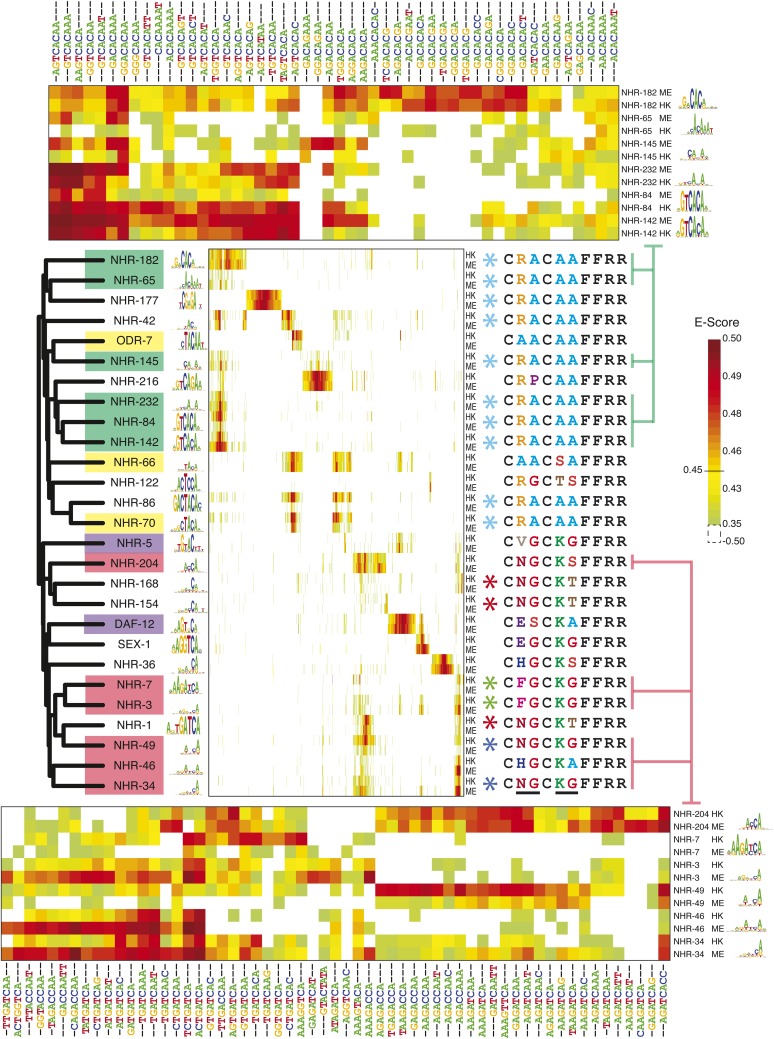
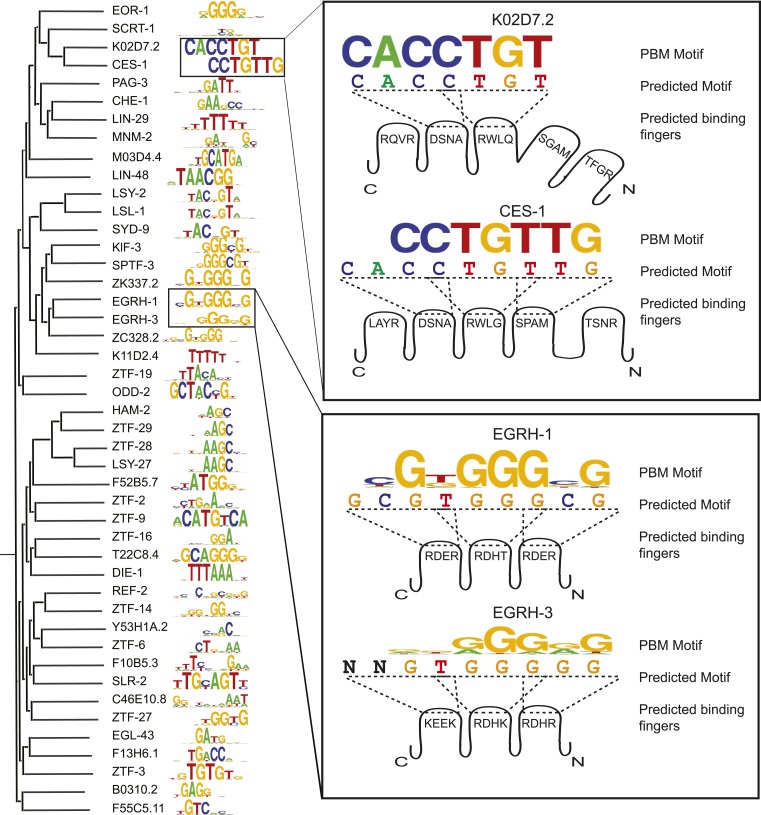
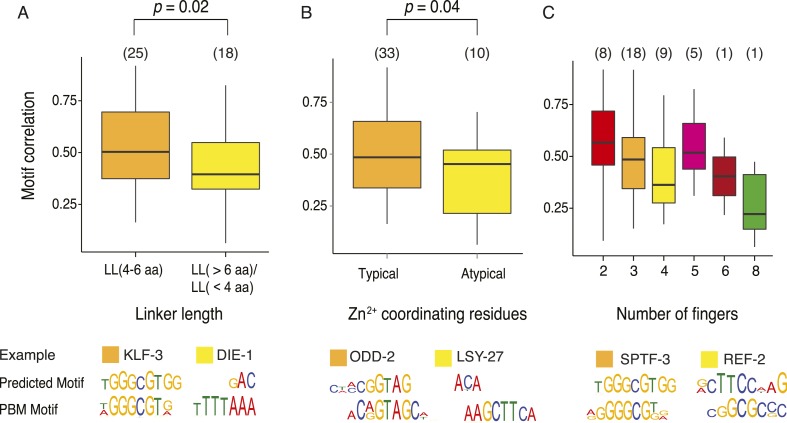
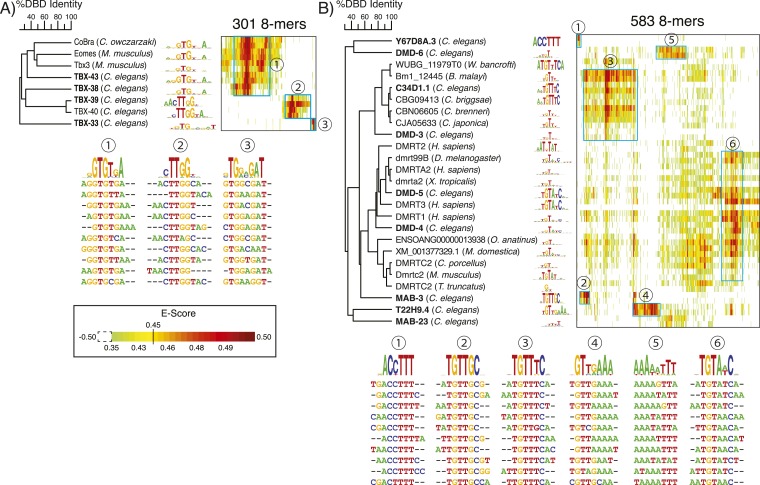
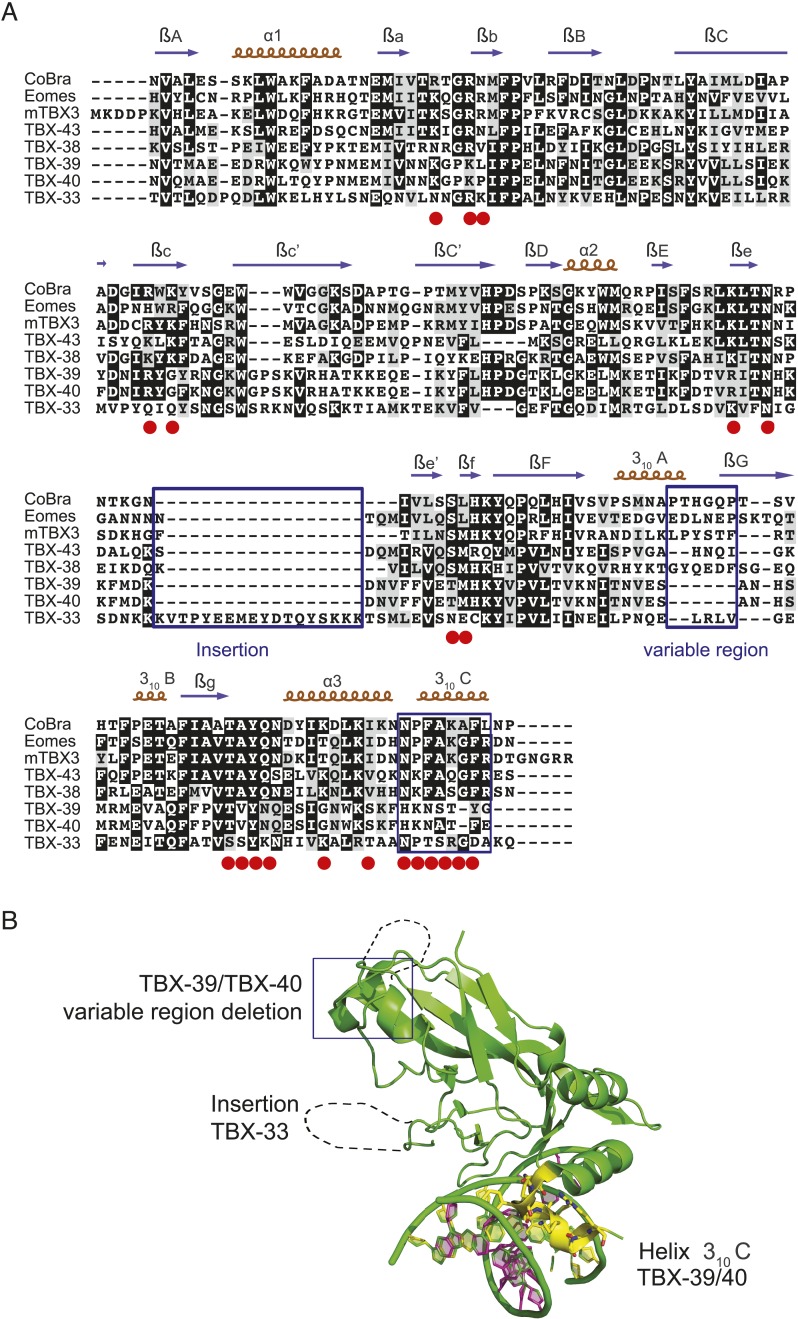
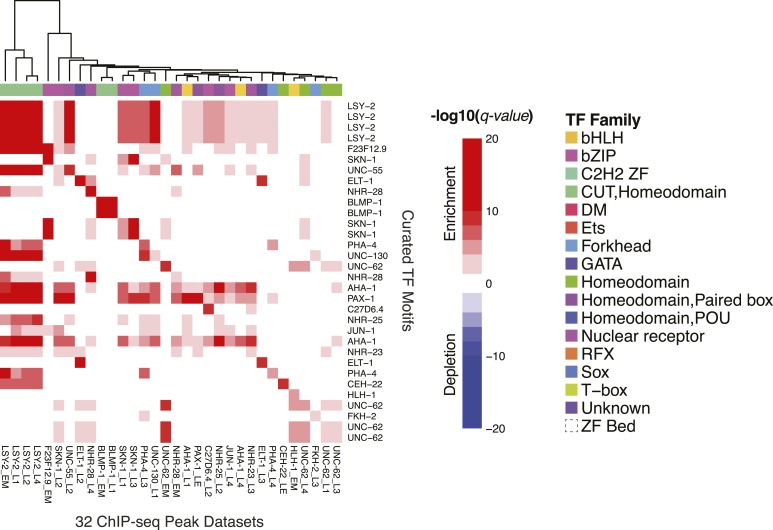
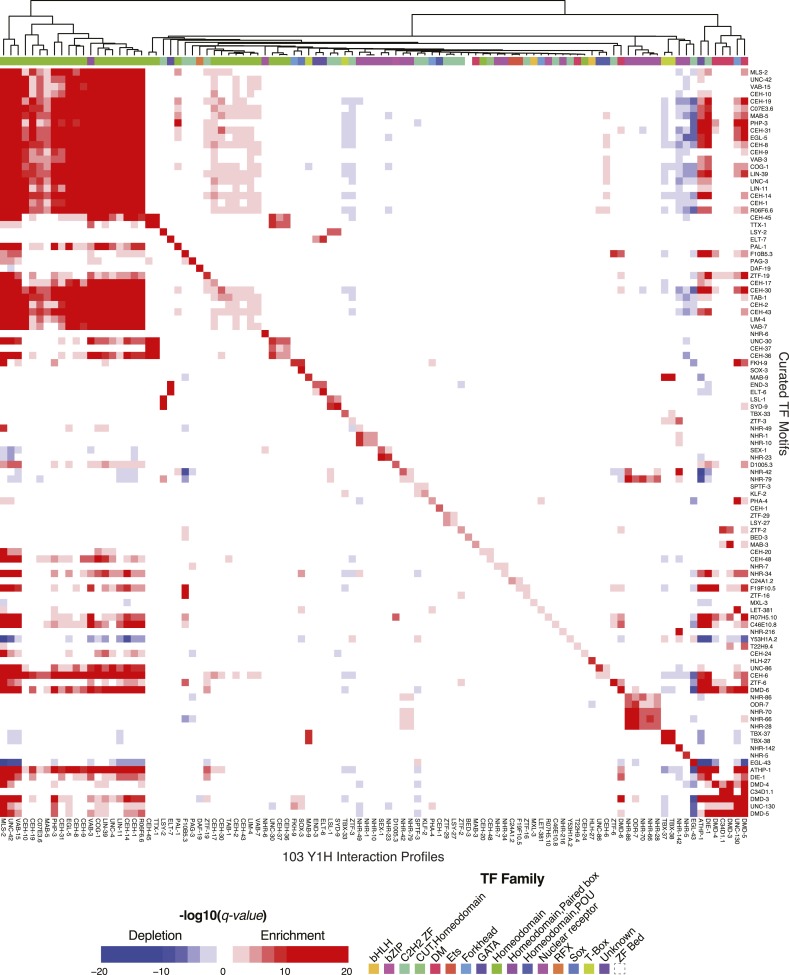
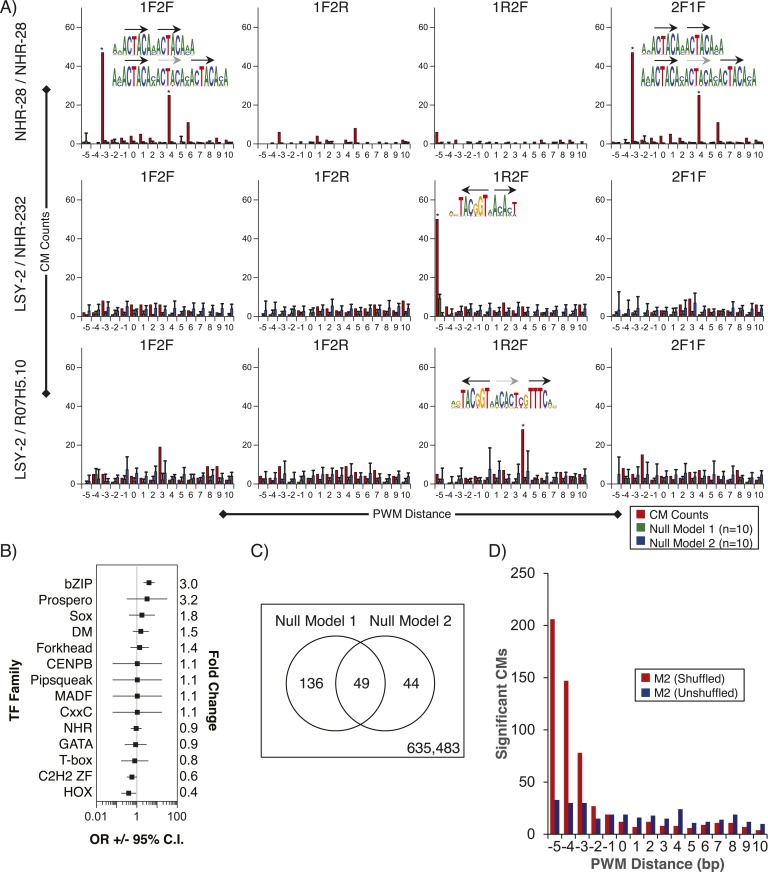
Caenorhabditis elegans is a powerful model for studying gene regulation, as it has a compact genome and a wealth of genomic tools. However, identification of regulatory elements has been limited, as DNA-binding motifs are known for only 71 of the estimated 763 sequence-specific transcription factors (TFs). To address this problem, we performed protein binding microarray experiments on representatives of canonical TF families in C. elegans, obtaining motifs for 129 TFs. Additionally, we predict motifs for many TFs that have DNA-binding domains similar to those already characterized, increasing coverage of binding specificities to 292 C. elegans TFs (∼40%). These data highlight the diversification of binding motifs for the nuclear hormone receptor and C2H2 zinc finger families and reveal unexpected diversity of motifs for T-box and DM families. Motif enrichment in promoters of functionally related genes is consistent with known biology and also identifies putative regulatory roles for unstudied TFs.
Keywords: C. elegans; DM; T-box; binding specificities; computational biology; evolutionary biology; genomics; nuclear hormone receptors; protein-binding microarray; systems biology; transcription factors.
Conflict of interest statement
The authors declare that no competing interests exist.
Figures

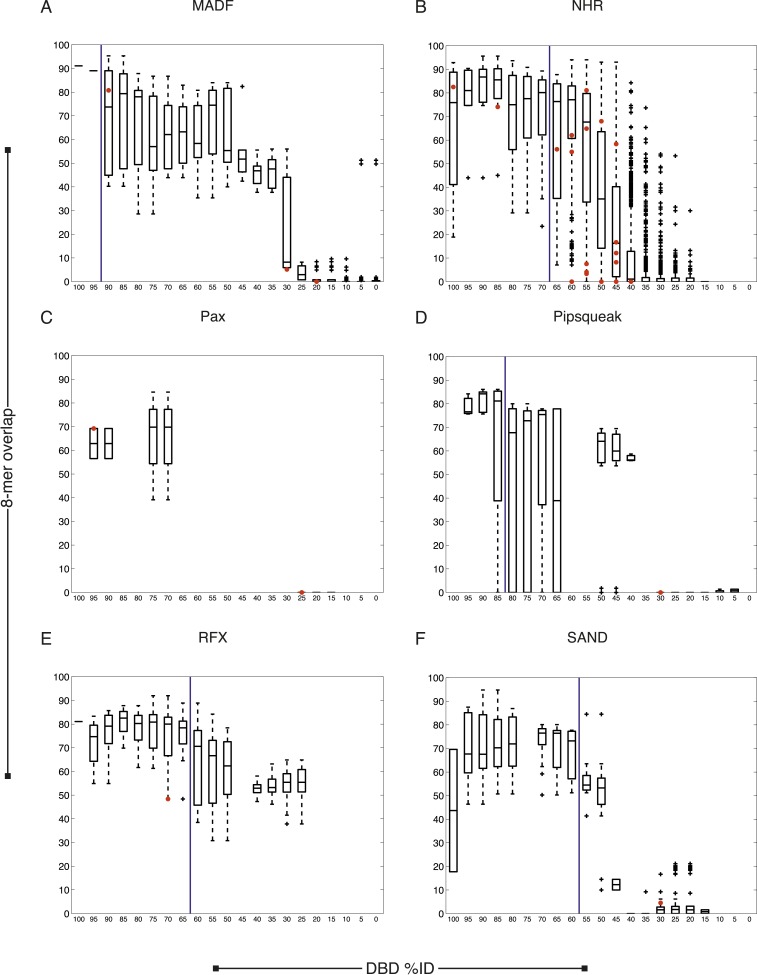
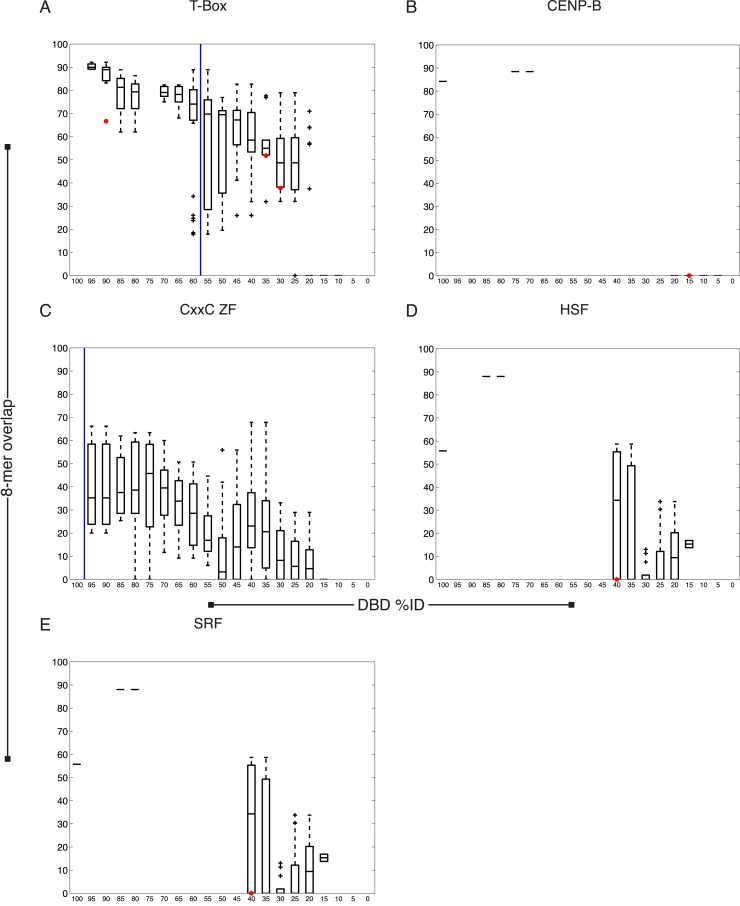
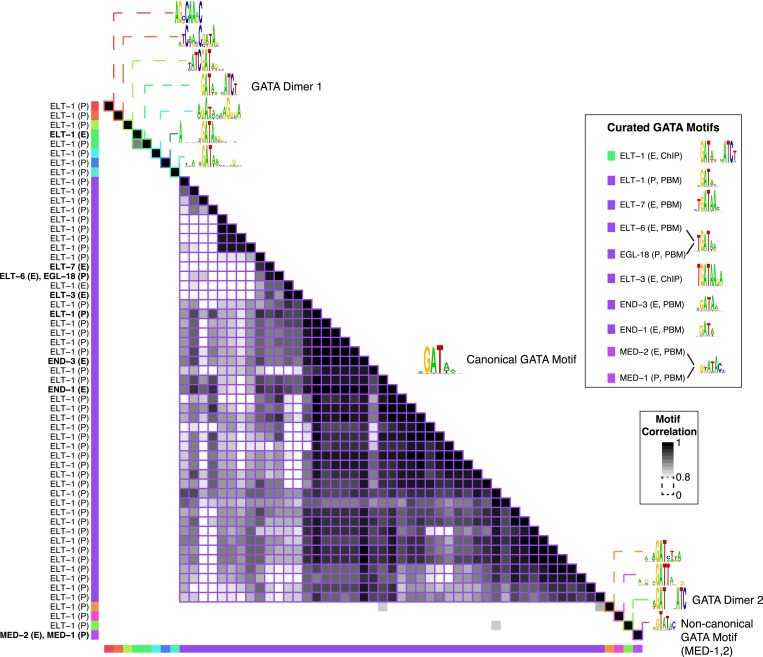
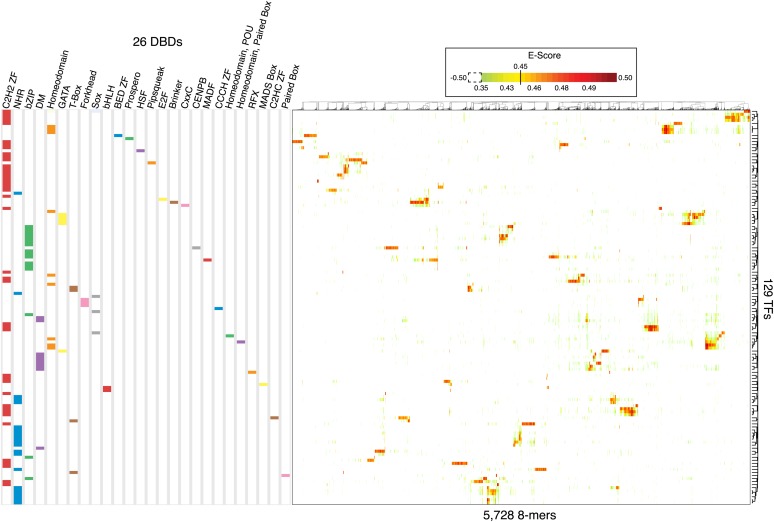
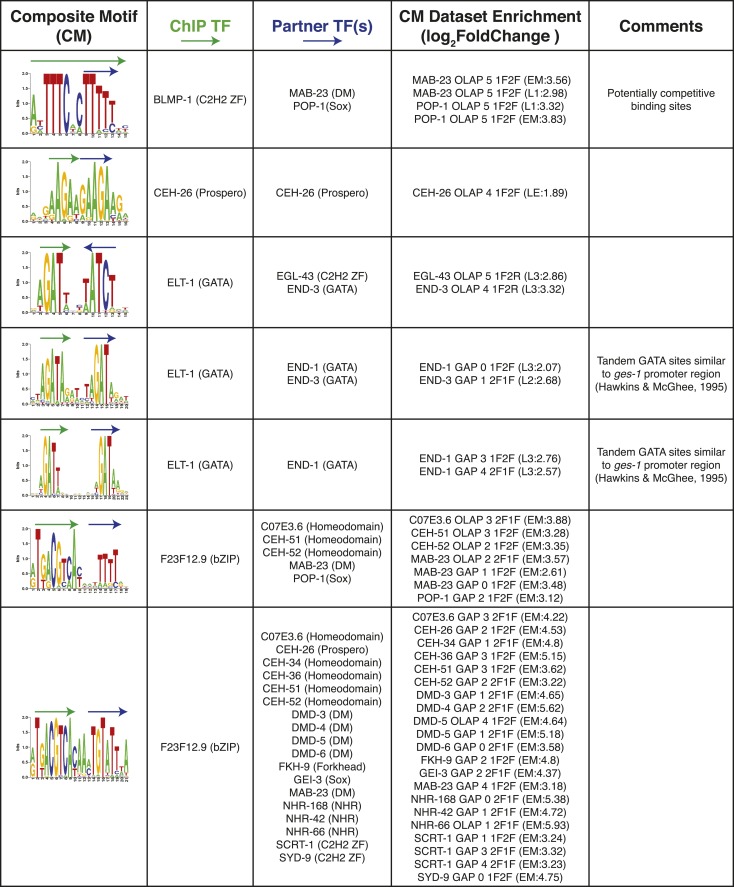
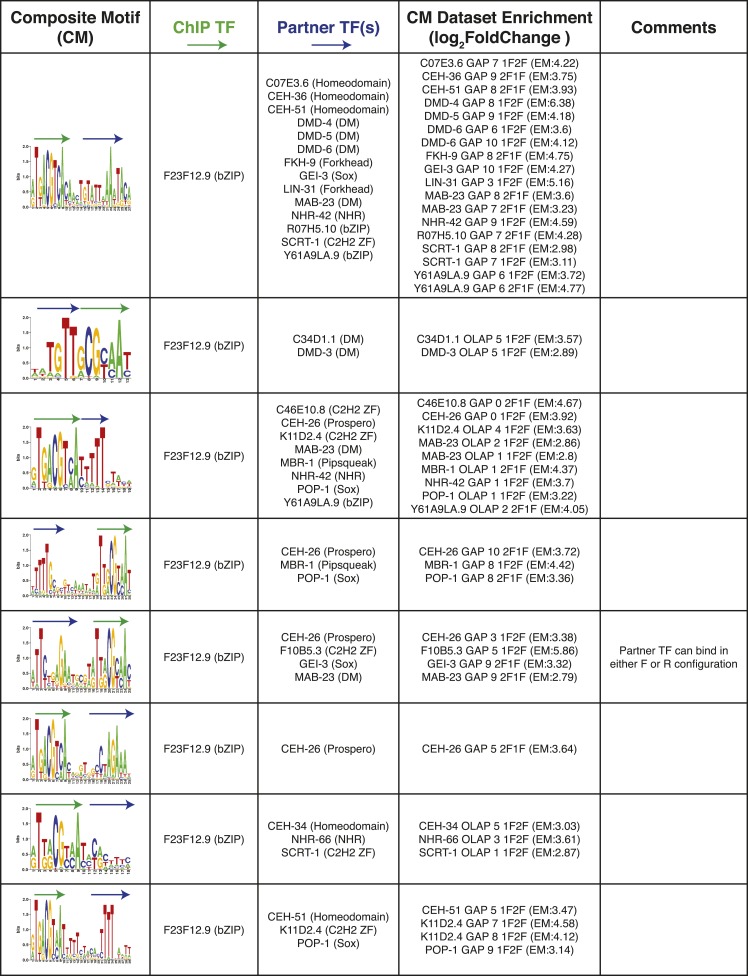
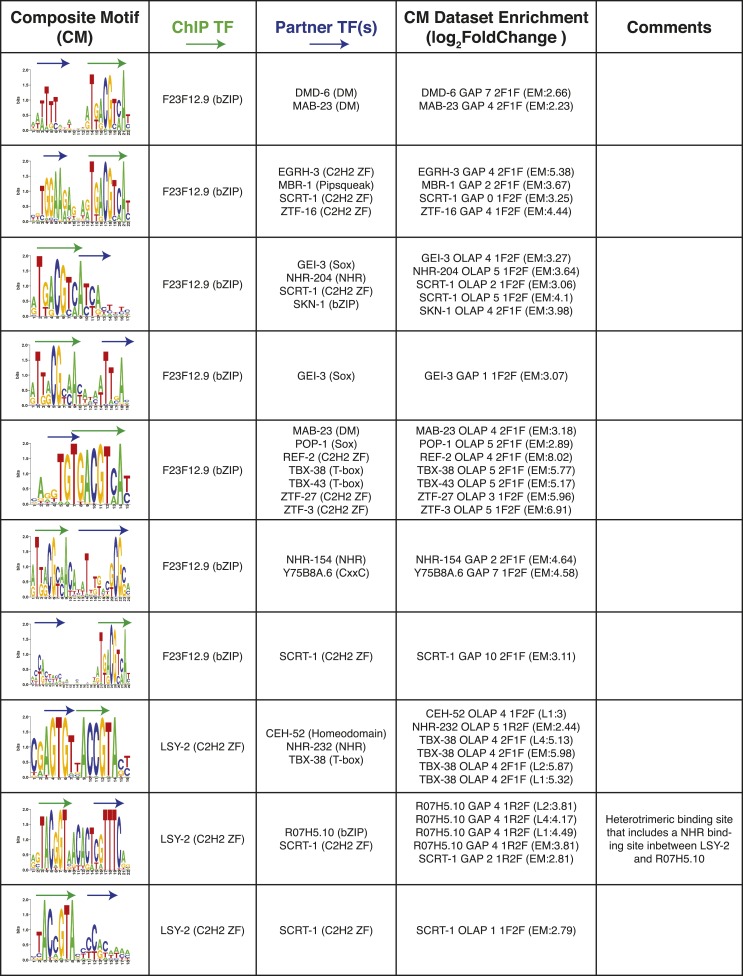
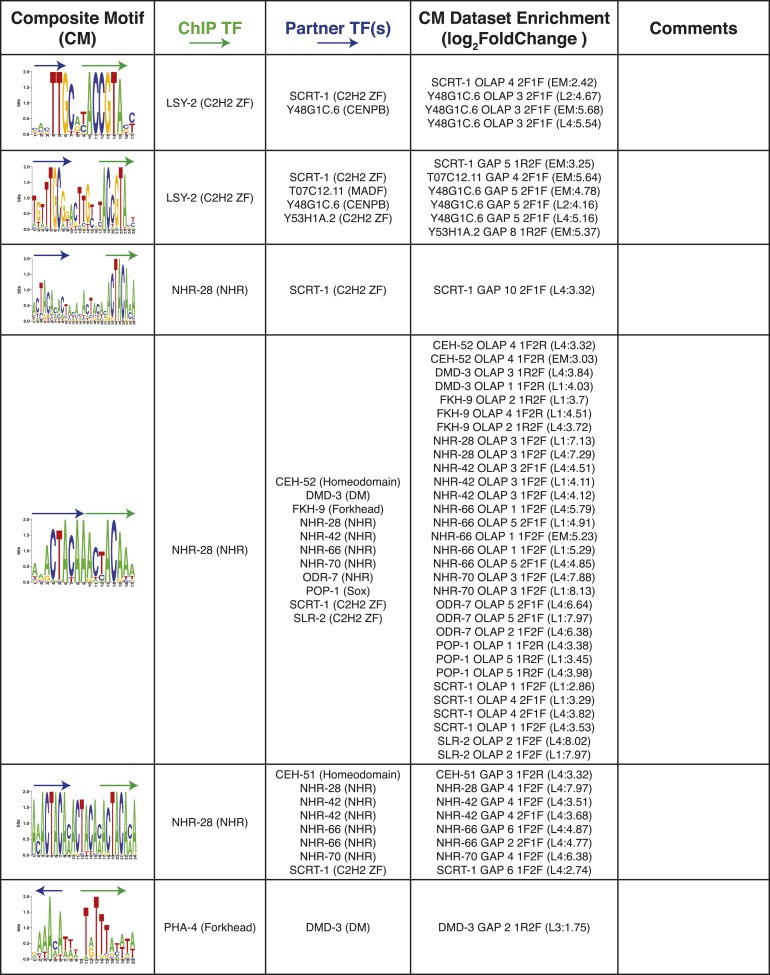
References
-
- Araya CL, Kawli T, Kundaje A, Jiang L, Wu B, Vafeados D, Terrell R, Weissdepp P, Gevirtzman L, Mace D. Niu W, Boyle AP, Xie D, Ma L, Murray JI, Reinke V, Waterston RH, Snyder M. Regulatory analysis of the C. elegans genome with spatiotemporal resolution. Nature. 2014;512:400–405. doi: 10.1038/nature13497. - DOI - PMC - PubMed
-
- Arda HE, Taubert S, MacNeil LT, Conine CC, Tsuda B, Van Gilst M, Sequerra R, Doucette-Stamm L, Yamamoto KR, Walhout AJ. Functional modularity of nuclear hormone receptors in a Caenorhabditis elegans metabolic gene regulatory network. Molecular Systems Biology. 2010;6:367. doi: 10.1038/msb.2010.23. - DOI - PMC - PubMed
-
- Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, Harris MA, Hill DP, Issel-Tarver L, Kasarskis A, Lewis S, Matese JC, Richardson JE, Ringwald M, Rubin GM, Sherlock G. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nature Genetics. 2000;25:25–29. doi: 10.1038/75556. - DOI - PMC - PubMed
-
- Badis G, Berger MF, Philippakis AA, Talukder S, Gehrke AR, Jaeger SA, Chan ET, Metzler G, Vedenko A, Chen X, Kuznetsov H, Wang CF, Coburn D, Newburger DE, Morris Q, Hughes TR, Bulyk ML. Diversity and complexity in DNA recognition by transcription factors. Science. 2009;324:1720–1723. doi: 10.1126/science.1162327. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
