

The power of gene-based rare variant methods to detect disease-associated variation and test hypotheses about complex disease
- PMID: 25906071
- PMCID: PMC4407972
- DOI: 10.1371/journal.pgen.1005165
The power of gene-based rare variant methods to detect disease-associated variation and test hypotheses about complex disease
Abstract
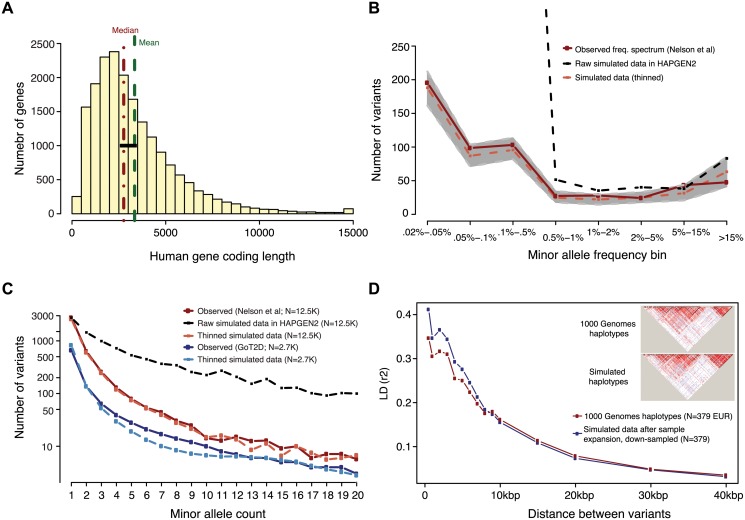
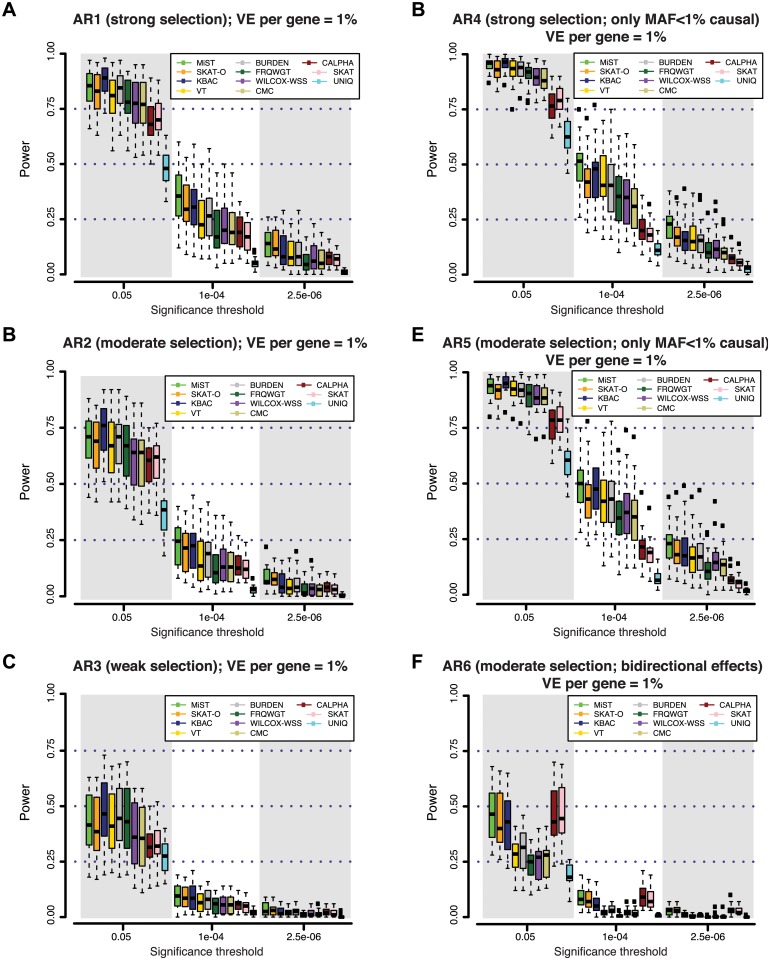
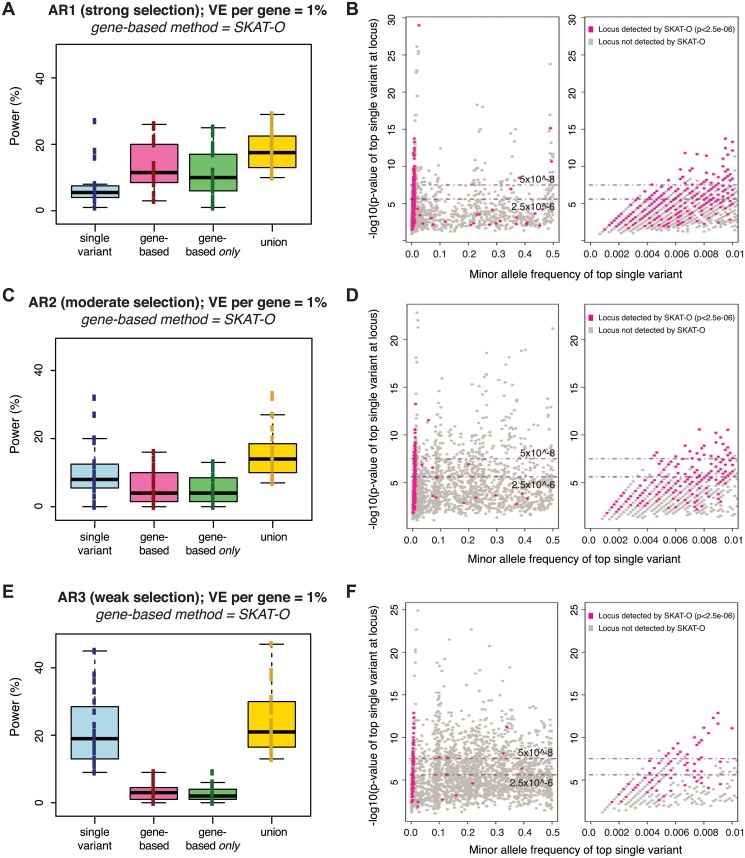
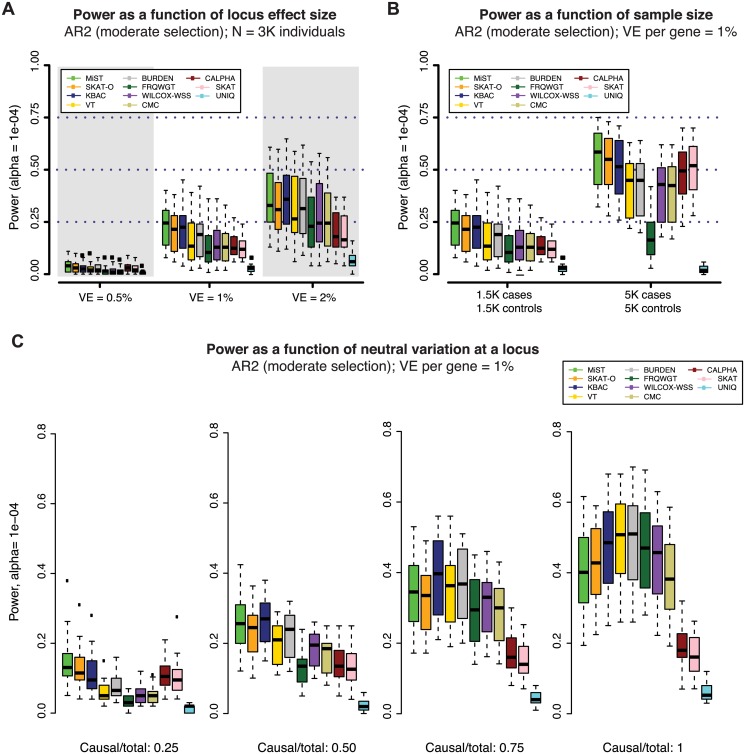
Genome and exome sequencing in large cohorts enables characterization of the role of rare variation in complex diseases. Success in this endeavor, however, requires investigators to test a diverse array of genetic hypotheses which differ in the number, frequency and effect sizes of underlying causal variants. In this study, we evaluated the power of gene-based association methods to interrogate such hypotheses, and examined the implications for study design. We developed a flexible simulation approach, using 1000 Genomes data, to (a) generate sequence variation at human genes in up to 10K case-control samples, and (b) quantify the statistical power of a panel of widely used gene-based association tests under a variety of allelic architectures, locus effect sizes, and significance thresholds. For loci explaining ~1% of phenotypic variance underlying a common dichotomous trait, we find that all methods have low absolute power to achieve exome-wide significance (~5-20% power at α = 2.5 × 10(-6)) in 3K individuals; even in 10K samples, power is modest (~60%). The combined application of multiple methods increases sensitivity, but does so at the expense of a higher false positive rate. MiST, SKAT-O, and KBAC have the highest individual mean power across simulated datasets, but we observe wide architecture-dependent variability in the individual loci detected by each test, suggesting that inferences about disease architecture from analysis of sequencing studies can differ depending on which methods are used. Our results imply that tens of thousands of individuals, extensive functional annotation, or highly targeted hypothesis testing will be required to confidently detect or exclude rare variant signals at complex disease loci.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
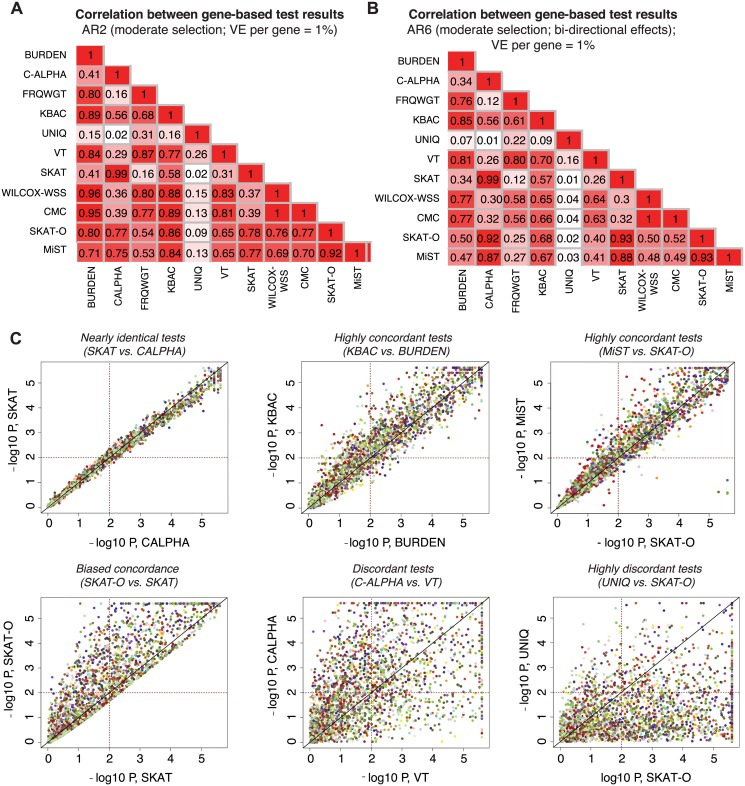
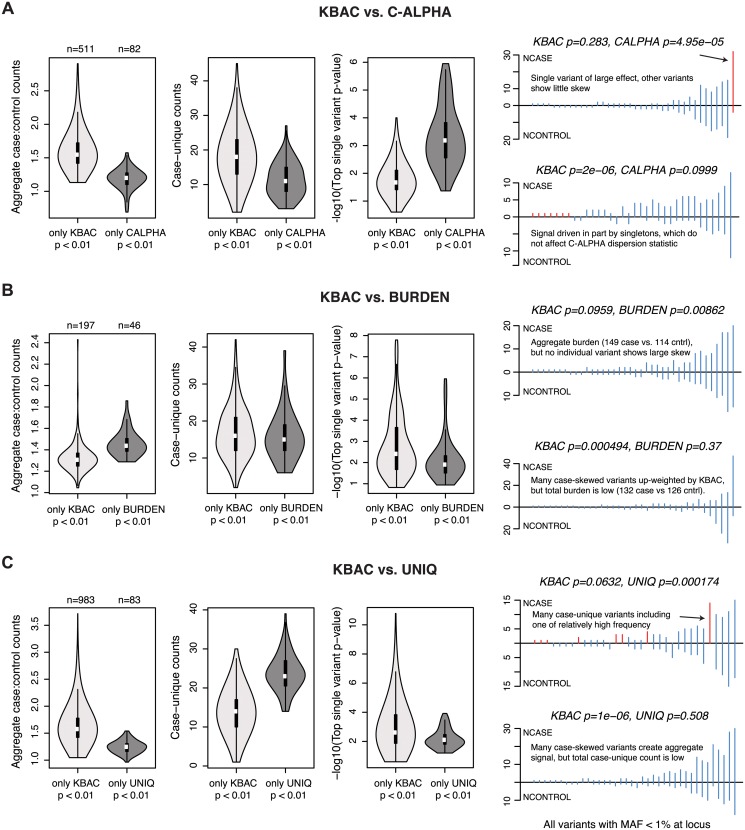
References
-
- Purcell S., Cherny S.S. & Sham P.C. Genetic Power Calculator: design of linkage and association genetic mapping studies of complex traits. Bioinformatics 19, 149–50 (2003). - PubMed
Publication types
MeSH terms
Grants and funding
- 098381/WT_/Wellcome Trust/United Kingdom
- 1RC2DK088389/DK/NIDDK NIH HHS/United States
- RC2-HG005688/HG/NHGRI NIH HHS/United States
- U01 DK085545/DK/NIDDK NIH HHS/United States
- R01 DK098032/DK/NIDDK NIH HHS/United States
- R01 HG000376/HG/NHGRI NIH HHS/United States
- T32 GM007753/GM/NIGMS NIH HHS/United States
- R56 HG000376/HG/NHGRI NIH HHS/United States
- R01 DK062370/DK/NIDDK NIH HHS/United States
- DK062370/DK/NIDDK NIH HHS/United States
- HG000376/HG/NHGRI NIH HHS/United States
- U01-DK085545/DK/NIDDK NIH HHS/United States
- R56 DK062370/DK/NIDDK NIH HHS/United States
- 090367/WT_/Wellcome Trust/United Kingdom
- R01-DK098032/DK/NIDDK NIH HHS/United States
- RC2 DK088389/DK/NIDDK NIH HHS/United States
- T32GM008313/GM/NIGMS NIH HHS/United States
- U01 DK062370/DK/NIDDK NIH HHS/United States
- 090532/WT_/Wellcome Trust/United Kingdom
- T32 GM008313/GM/NIGMS NIH HHS/United States
- P30 DK020572/DK/NIDDK NIH HHS/United States
- T32GM007753/GM/NIGMS NIH HHS/United States
- WT_/Wellcome Trust/United Kingdom
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
