

Enhanced ligand sampling for relative protein-ligand binding free energy calculations
- PMID: 25906170
- PMCID: PMC4442669
- DOI: 10.1021/acs.jpcb.5b02348
Enhanced ligand sampling for relative protein-ligand binding free energy calculations
Abstract
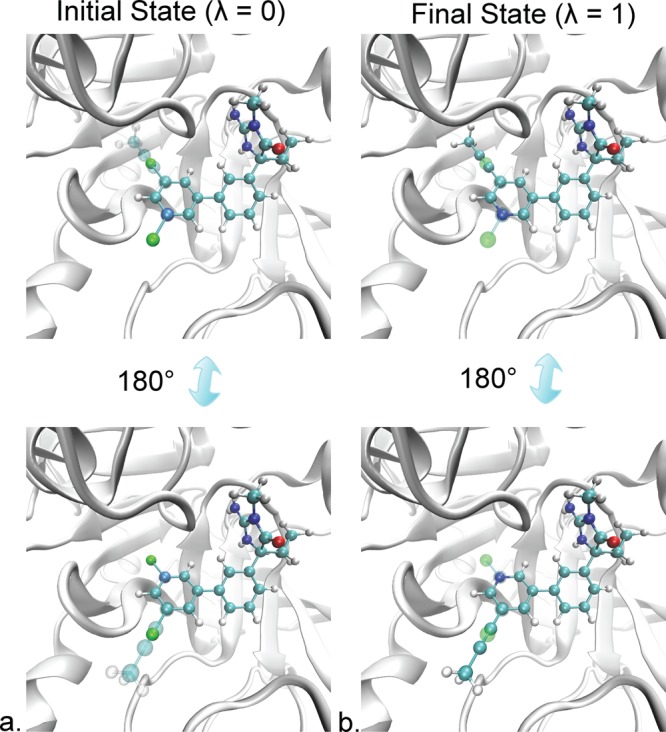
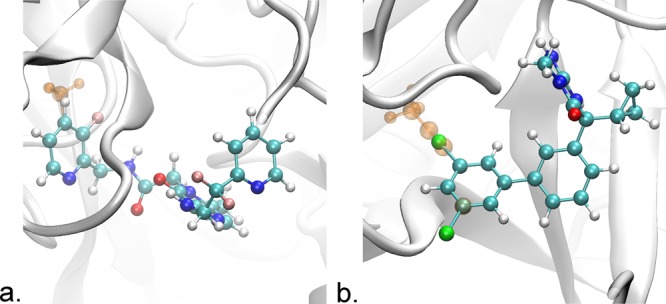
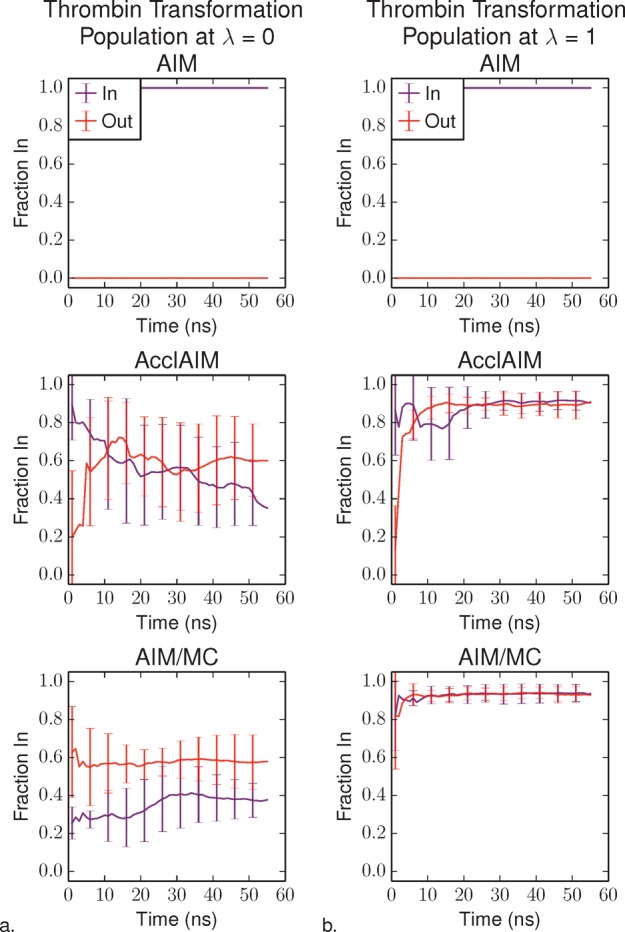
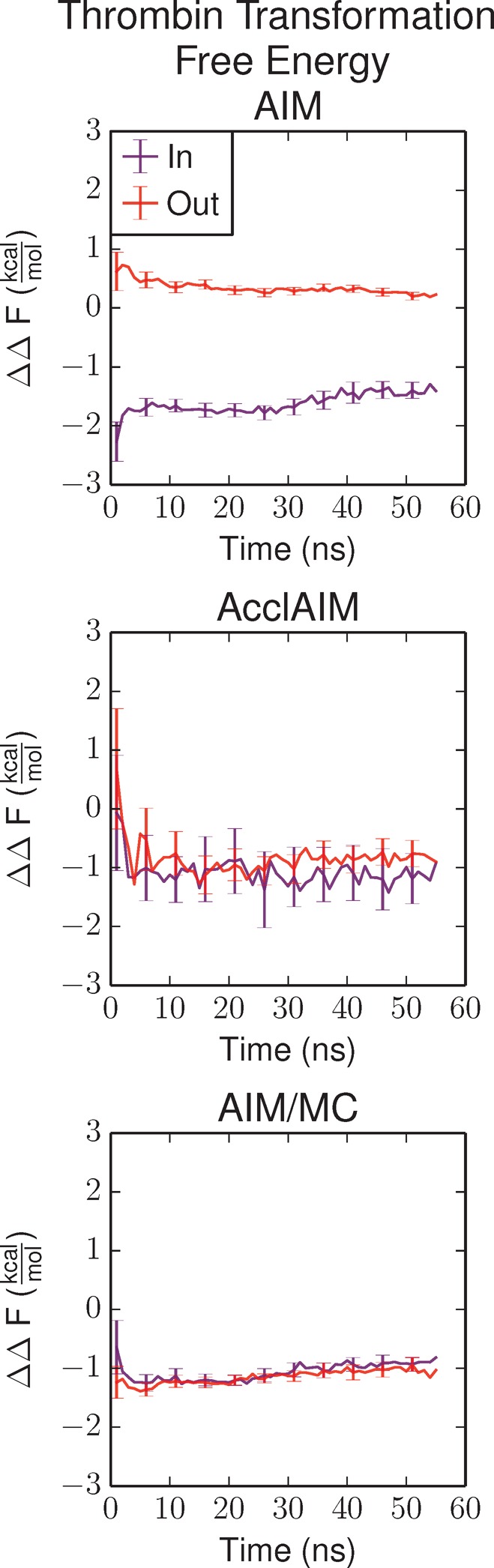
Free energy calculations are used to study how strongly potential drug molecules interact with their target receptors. The accuracy of these calculations depends on the accuracy of the molecular dynamics (MD) force field as well as proper sampling of the major conformations of each molecule. However, proper sampling of ligand conformations can be difficult when there are large barriers separating the major ligand conformations. An example of this is for ligands with an asymmetrically substituted phenyl ring, where the presence of protein loops hinders the proper sampling of the different ring conformations. These ring conformations become more difficult to sample when the size of the functional groups attached to the ring increases. The Adaptive Integration Method (AIM) has been developed, which adaptively changes the alchemical coupling parameter λ during the MD simulation so that conformations sampled at one λ can aid sampling at the other λ values. The Accelerated Adaptive Integration Method (AcclAIM) builds on AIM by lowering potential barriers for specific degrees of freedom at intermediate λ values. However, these methods may not work when there are very large barriers separating the major ligand conformations. In this work, we describe a modification to AIM that improves sampling of the different ring conformations, even when there is a very large barrier between them. This method combines AIM with conformational Monte Carlo sampling, giving improved convergence of ring populations and the resulting free energy. This method, called AIM/MC, is applied to study the relative binding free energy for a pair of ligands that bind to thrombin and a different pair of ligands that bind to aspartyl protease β-APP cleaving enzyme 1 (BACE1). These protein-ligand binding free energy calculations illustrate the improvements in conformational sampling and the convergence of the free energy compared to both AIM and AcclAIM.
Figures
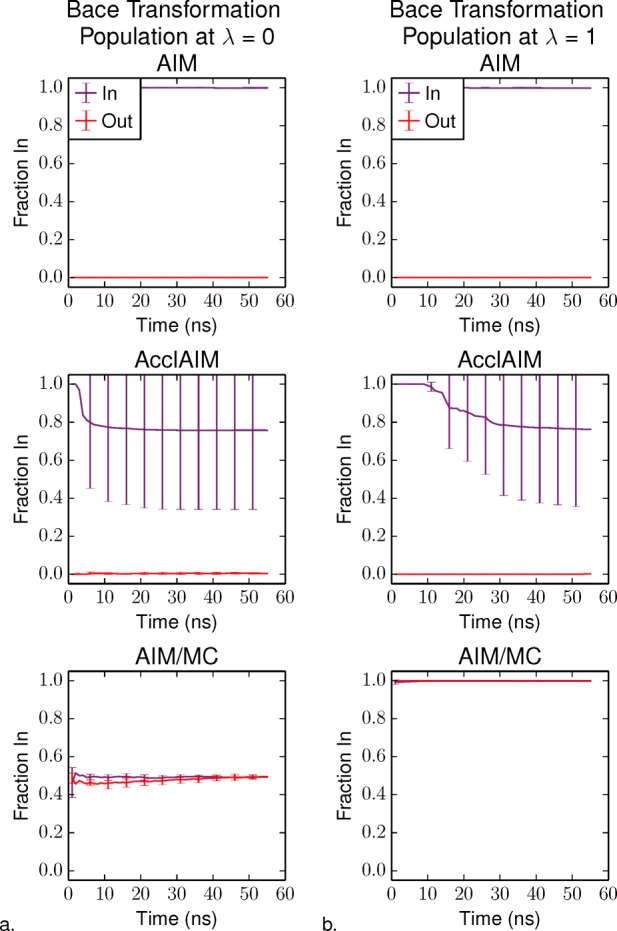
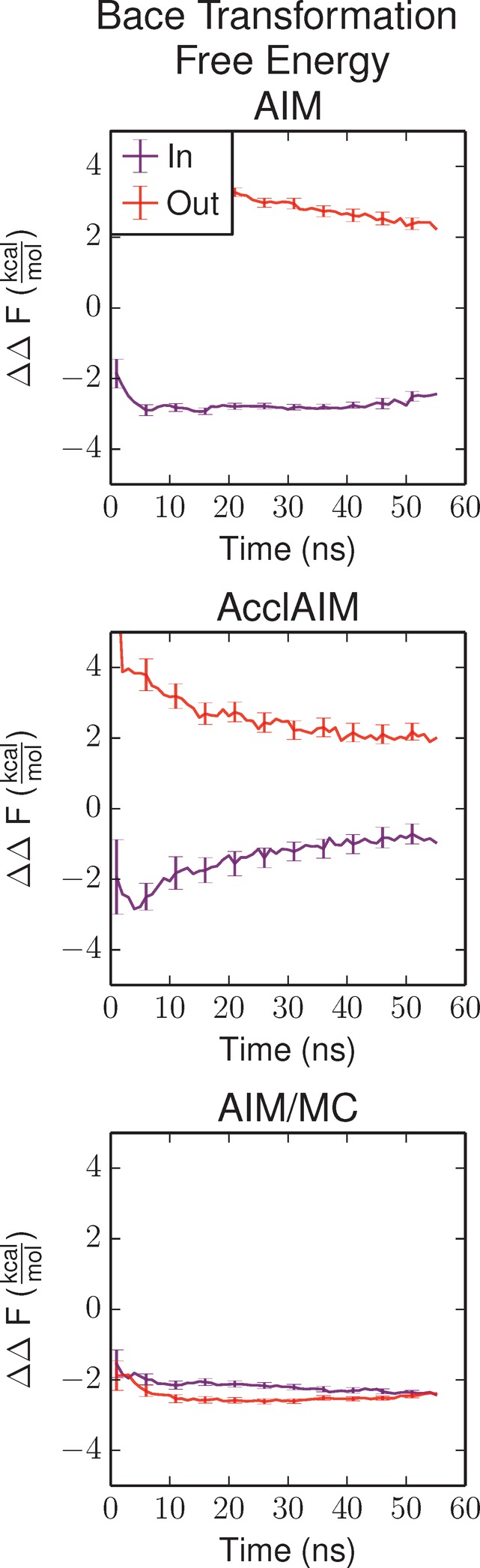
References
-
- Fasnacht M.; Swendsen R.; Rosenberg J. Adaptive Integration Method for Monte Carlo Simulations. Phys. Rev. E 2004, 69, 056704. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
