

G-CSF Promotes Neuroblastoma Tumorigenicity and Metastasis via STAT3-Dependent Cancer Stem Cell Activation
- PMID: 25908586
- PMCID: PMC4470771
- DOI: 10.1158/0008-5472.CAN-14-2946
G-CSF Promotes Neuroblastoma Tumorigenicity and Metastasis via STAT3-Dependent Cancer Stem Cell Activation
Abstract
Increasing evidence suggests that inflammatory cytokines play a critical role in tumor initiation and progression. A cancer stem cell (CSC)-like subpopulation in neuroblastoma is known to be marked by expression of the G-CSF receptor (G-CSFR). Here, we report on the mechanistic contributions of the G-CSFR in neuroblastoma CSCs. Specifically, we demonstrate that the receptor ligand G-CSF selectively activates STAT3 within neuroblastoma CSC subpopulations, promoting their expansion in vitro and in vivo. Exogenous G-CSF enhances tumor growth and metastasis in human xenograft and murine neuroblastoma tumor models. In response to G-CSF, STAT3 acts in a feed-forward loop to transcriptionally activate the G-CSFR and sustain neuroblastoma CSCs. Blockade of this G-CSF-STAT3 signaling loop with either anti-G-CSF antibody or STAT3 inhibitor depleted the CSC subpopulation within tumors, driving correlated tumor growth inhibition, decreased metastasis, and increased chemosensitivity. Taken together, our results define G-CSF as a CSC-activating factor in neuroblastoma, suggest a comprehensive reevaluation of the clinical use of G-CSF in these patients to support white blood cell counts, and suggest that direct targeting of the G-CSF-STAT3 signaling represents a novel therapeutic approach for neuroblastoma.
©2015 American Association for Cancer Research.
Conflict of interest statement
Figures
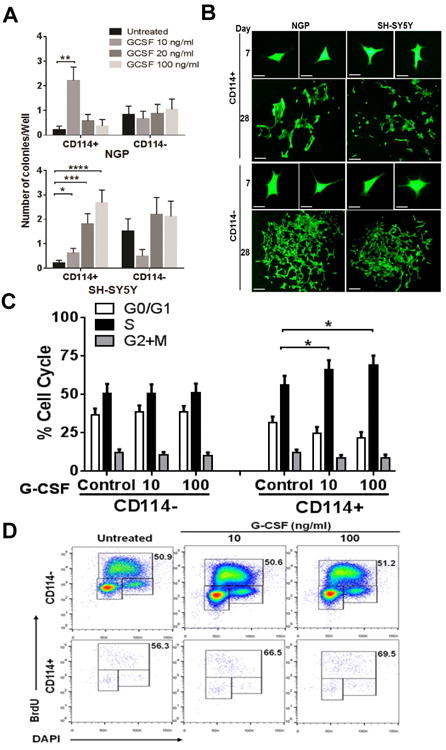
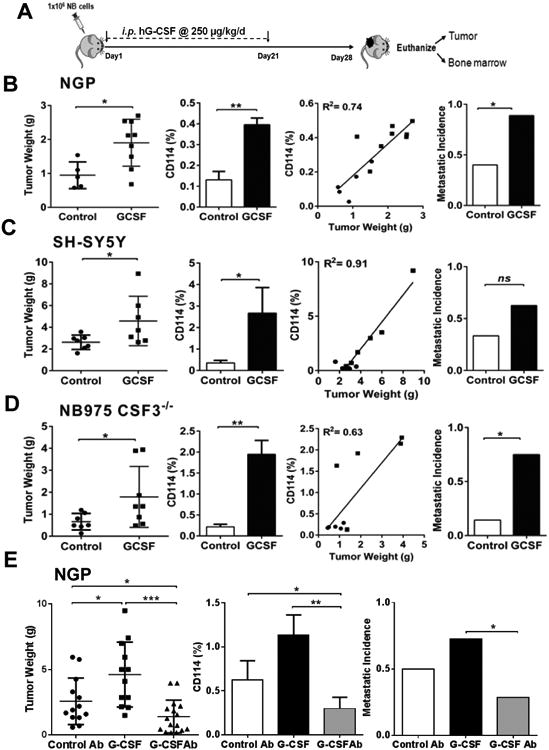
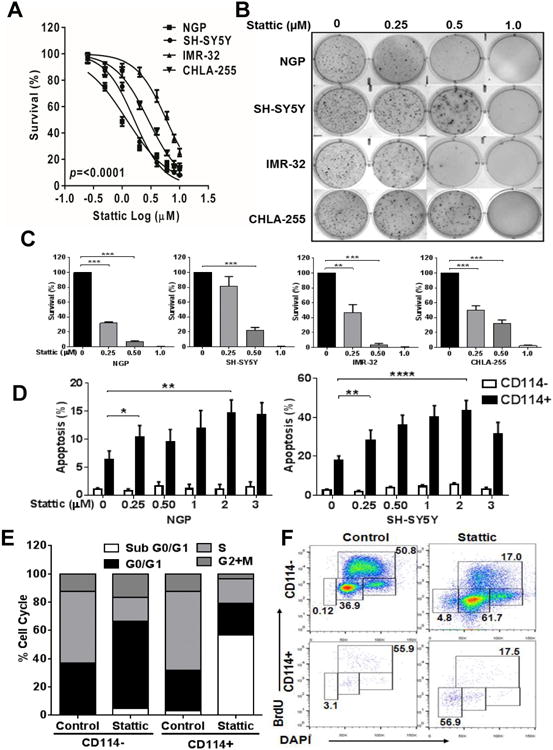
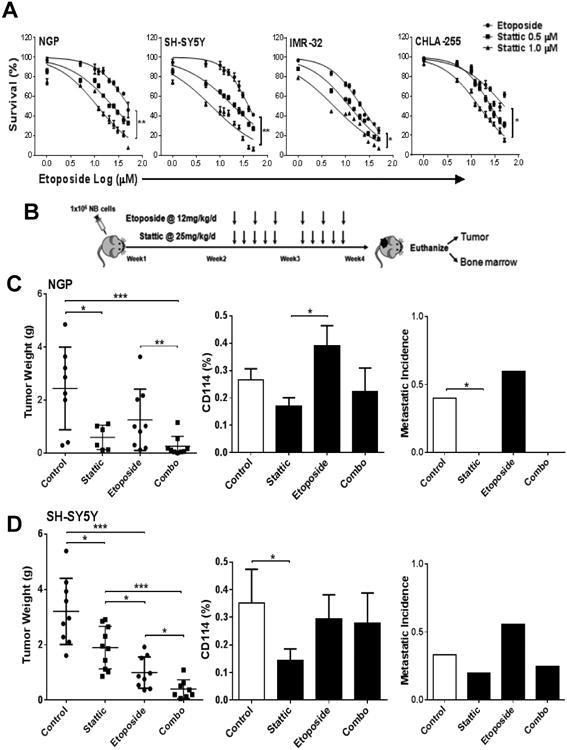
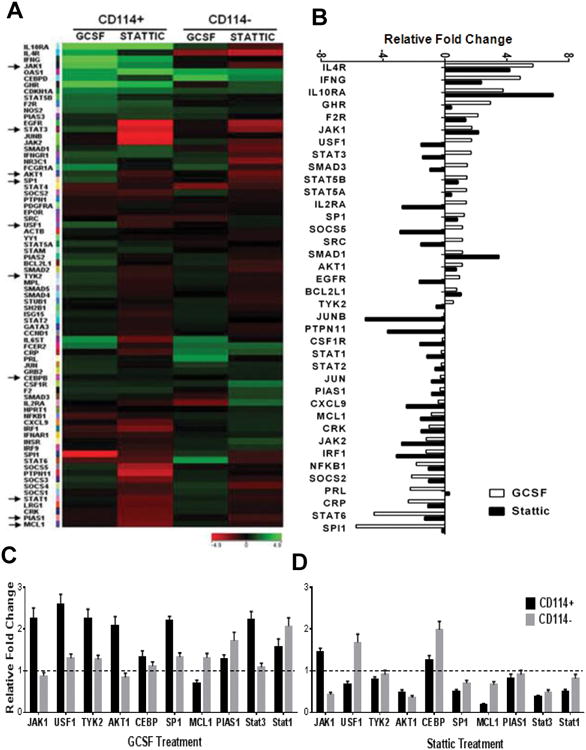
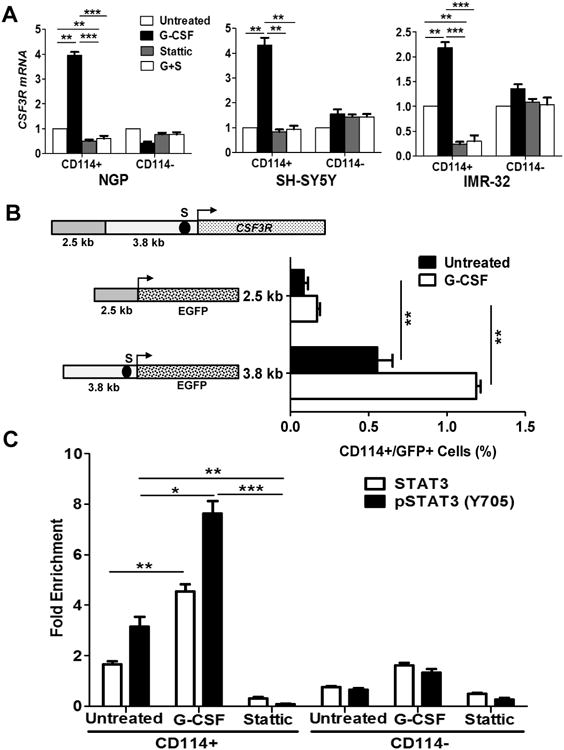
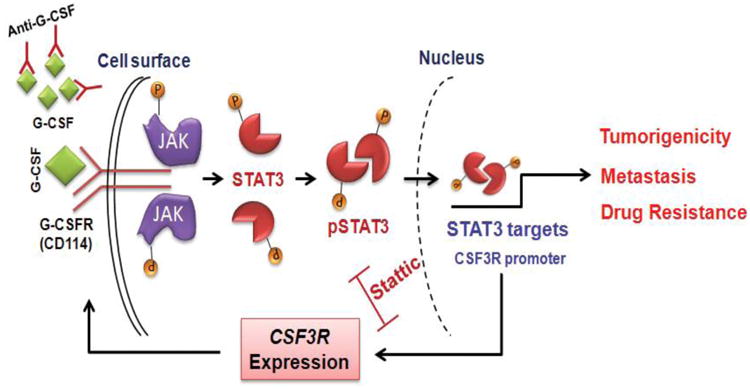
Comment in
-
G-CSF Is a Cancer Stem Cell-Specific Growth Factor-Letter.Cancer Res. 2015 Sep 15;75(18):3991. doi: 10.1158/0008-5472.CAN-15-1445. Epub 2015 Sep 3. Cancer Res. 2015. PMID: 26337906 No abstract available.
-
G-CSF Is a Cancer Stem Cell-Specific Growth Factor-Response.Cancer Res. 2015 Sep 15;75(18):3992. doi: 10.1158/0008-5472.CAN-15-1599. Epub 2015 Sep 3. Cancer Res. 2015. PMID: 26337907 Free PMC article. No abstract available.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
