

Loss of PTPRM associates with the pathogenic development of colorectal adenoma-carcinoma sequence
- PMID: 25910225
- PMCID: PMC5386118
- DOI: 10.1038/srep09633
Loss of PTPRM associates with the pathogenic development of colorectal adenoma-carcinoma sequence
Abstract
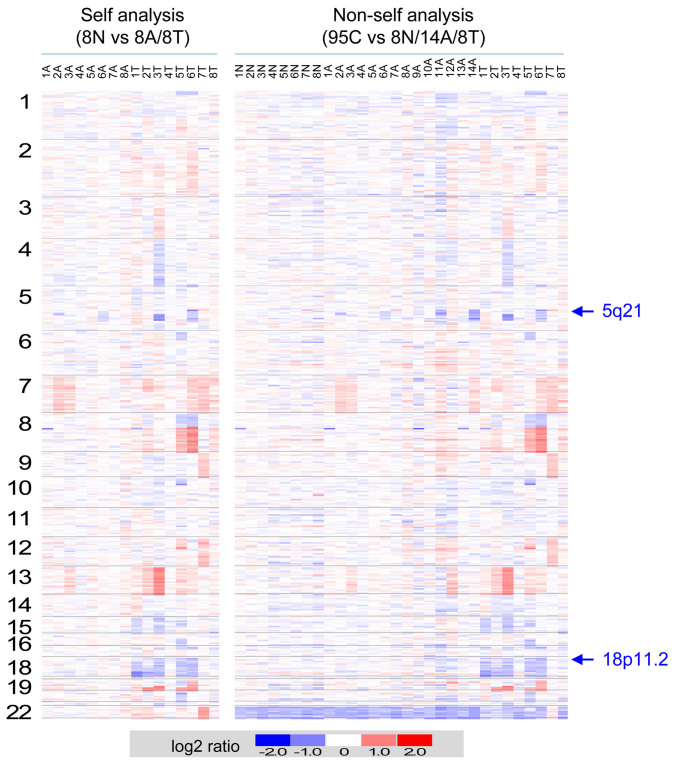
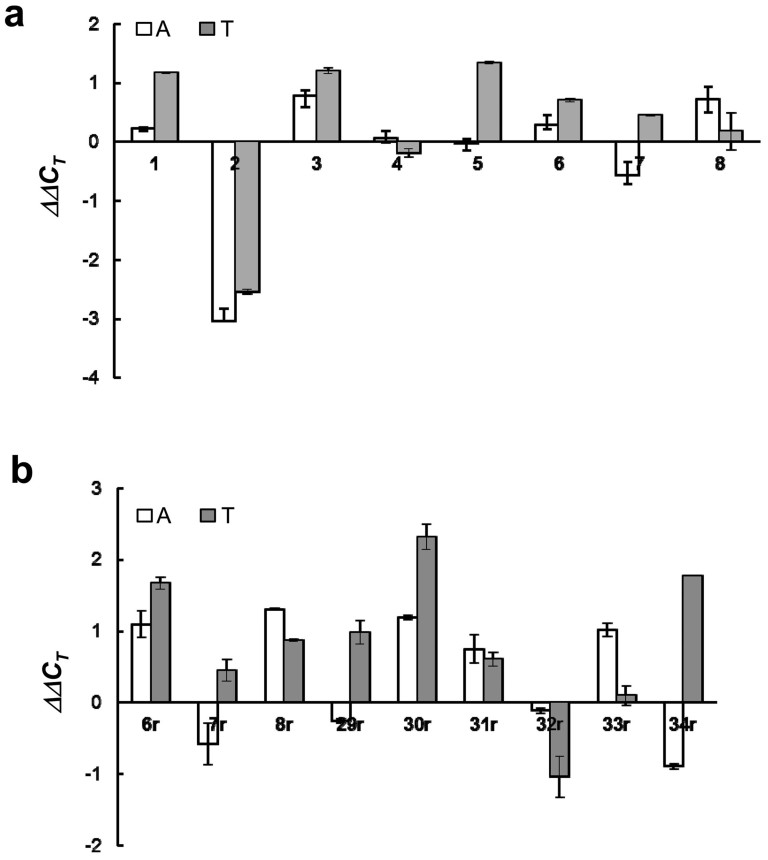
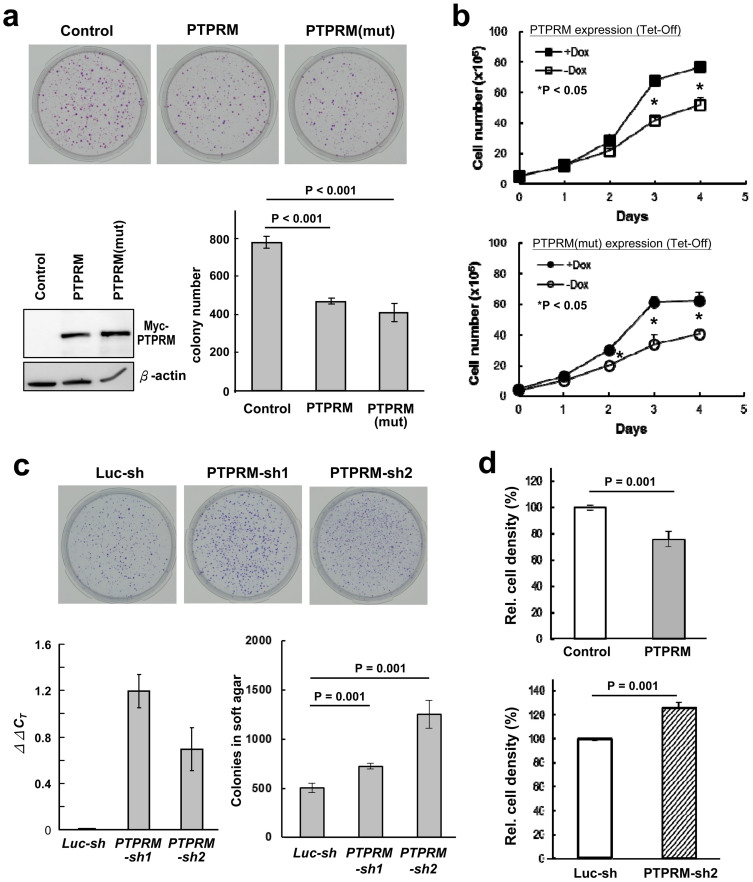
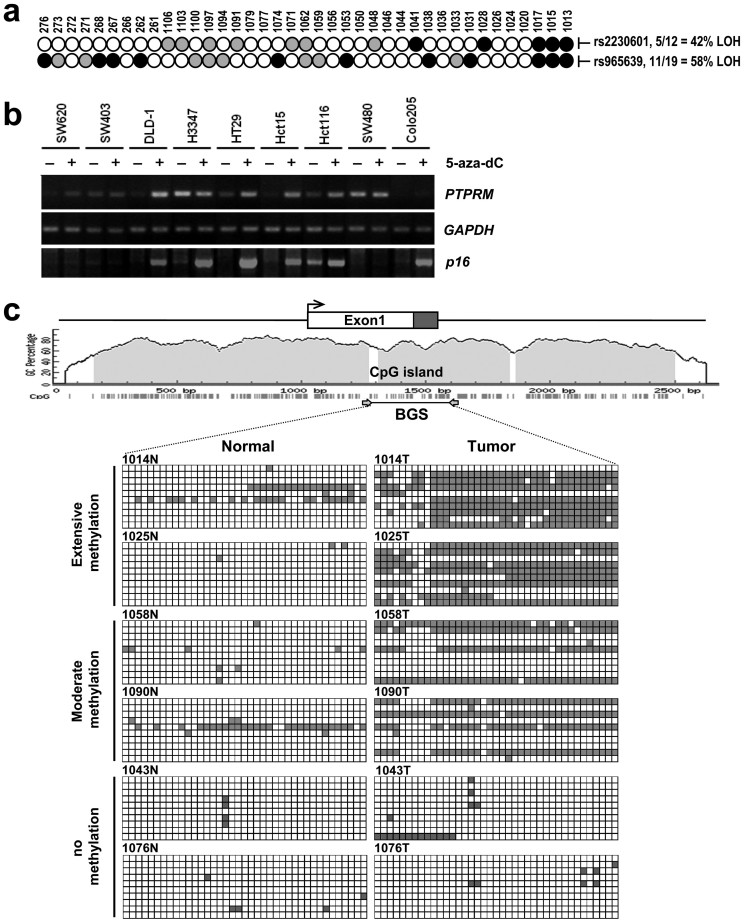
Identification and functional analysis of genes from genetically altered chromosomal regions would suggest new molecular targets for cancer diagnosis and treatment. Here we performed a genome-wide analysis of chromosomal copy number alterations (CNAs) in matching sets of colon mucosa-adenoma-carcinoma samples using high-throughput oligonucleotide microarray analysis. In silico analysis of NCBI GEO and TCGA datasets allowed us to uncover the significantly altered genes (p ≤ 0.001) associated with the identified CNAs. We performed quantitative PCR analysis of the genomic and complementary DNA derived from primary mucosa, adenoma, and carcinoma samples, and confirmed the recurrent loss and down-regulation of PTPRM in colon adenomas and carcinomas. Functional characterization demonstrated that PTPRM negatively regulates cell growth and colony formation, whereas loss of PTPRM promotes oncogenic cell growth. We further showed that, in accordance to Knudson's two-hit hypothesis, inactivation of PTPRM in colon cancer was mainly attributed to loss of heterozygosity and promoter hypermethylation. Taken together, this study demonstrates a putative tumor suppressive role for PTPRM and that genetic and epigenetic alterations of PTPRM may contribute to early step of colorectal tumorigenesis.
Conflict of interest statement
The authors declare no competing financial interests.
Figures

References
-
- Fearon E. R. & Vogelstein B. A genetic model for colorectal tumorigenesis. Cell 61, 759–767 (1990). - PubMed
-
- Kinzler K. W. & Vogelstein B. Lessons from hereditary colorectal cancer. Cell 87, 159–170 (1996). - PubMed
-
- Leslie A., Carey F. A., Pratt N. R. & Steele R. J. The colorectal adenoma-carcinoma sequence. Br J Surg 89, 845–860 (2002). - PubMed
-
- Vogelstein B. et al. Genetic alterations during colorectal-tumor development. N Engl J Med 319, 525–532 (1988). - PubMed
-
- Habermann J. K. et al. Stage-specific alterations of the genome, transcriptome, and proteome during colorectal carcinogenesis. Genes Chromosomes Cancer 46, 10–26 (2007). - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
