

Identification of rare protein disulfide isomerase gene variants in amyotrophic lateral sclerosis patients
- PMID: 25913742
- PMCID: PMC5553116
- DOI: 10.1016/j.gene.2015.04.035
Identification of rare protein disulfide isomerase gene variants in amyotrophic lateral sclerosis patients
Abstract
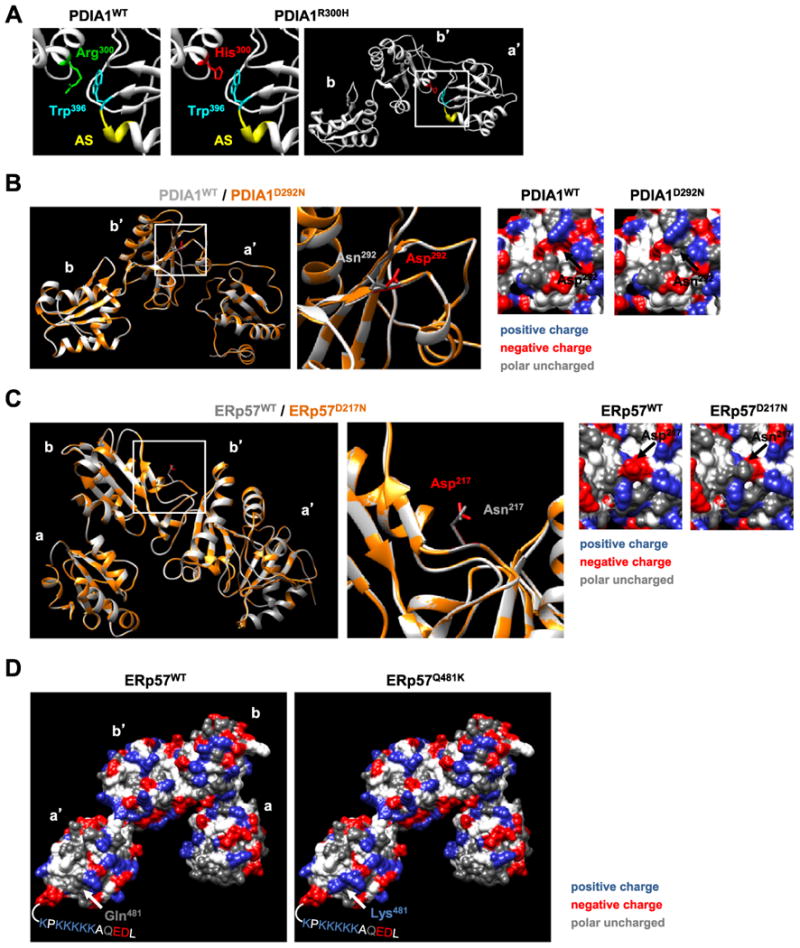
Disruption of endoplasmic reticulum (ER) proteostasis is a salient feature of amyotrophic lateral sclerosis (ALS). Upregulation of ER foldases of the protein disulfide isomerase (PDI) family has been reported in ALS mouse models and spinal cord tissue and body fluids derived from sporadic ALS cases. Although in vitro studies suggest a neuroprotective role of PDIs in ALS, the possible contribution of genetic mutations of these ER foldases in the disease process remains unknown. Interestingly, intronic variants of the PDIA1 gene were recently reported as a risk factor for ALS. Here, we initially screened for mutations in two major PDI genes (PDIA1/P4HB and PDIA3/ERp57) in a US cohort of 96 familial and 96 sporadic ALS patients using direct DNA sequencing. Then, 463 familial and 445 sporadic ALS patients from two independent cohorts were also screened for mutations in these two genes using whole exome sequencing. A total of nine PDIA1 missense variants and seven PDIA3 missense variants were identified in 16 ALS patients. We have identified several novel and rare single nucleotide polymorphisms (SNPs) in both genes that are enriched in ALS cases compared with a large group of control subjects showing a frequency of around 1% in ALS cases. The possible biological and structural impact of these ALS-linked PDI variants is also discussed.
Keywords: Amyotrophic lateral sclerosis; ER stress; ERp57; PDIA1; Protein disulfide isomerase.
Copyright © 2015 Elsevier B.V. All rights reserved.
Conflict of interest statement
The authors declare no conflict of interest.
Figures

References
-
- Andreu CI, Woehlbier U, Torres M, Hetz C. Protein disulfide isomerases in neurodegeneration: from disease mechanisms to biomedical applications. FEBS Lett. 2012;586:2826–2834. - PubMed
-
- Atkin JD, Farg MA, Turner BJ, Tomas D, Lysaght JA, Nunan J, Rembach A, Nagley P, Beart PM, Cheema SS, Horne MK. Induction of the unfolded protein response in familial amyotrophic lateral sclerosis and association of protein-disulfide isomerase with superoxide dismutase 1. J Biol Chem. 2006;281:30152–30165. - PubMed
-
- Atkin JD, Farg MA, Walker AK, McLean C, Tomas D, Horne MK. Endoplasmic reticulum stress and induction of the unfolded protein response in human sporadic amyotrophic lateral sclerosis. Neurobiol Dis. 2008;30:400–407. - PubMed
-
- Atkin JD, Farg MA, Soo KY, Walker AK, Halloran M, Turner BJ, Nagley P, Horne MK. Mutant SOD1 inhibits ER-Golgi transport in amyotrophic lateral sclerosis. J Neurochem. 2014;129:190–204. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- UC2 HL103010/HL/NHLBI NIH HHS/United States
- RC2 HL102926/HL/NHLBI NIH HHS/United States
- R01 NS073873/NS/NINDS NIH HHS/United States
- RC2 HL102924/HL/NHLBI NIH HHS/United States
- RC2 HL103010/HL/NHLBI NIH HHS/United States
- R01 NS050557/NS/NINDS NIH HHS/United States
- RC2 HL102923/HL/NHLBI NIH HHS/United States
- UC2 HL102926/HL/NHLBI NIH HHS/United States
- UC2 HL102923/HL/NHLBI NIH HHS/United States
- UC2 HL102924/HL/NHLBI NIH HHS/United States
- RC2 HL102925/HL/NHLBI NIH HHS/United States
- UC2 HL102925/HL/NHLBI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
