

Lipopolysaccharide (LPS)-Induced Biliary Epithelial Cell NRas Activation Requires Epidermal Growth Factor Receptor (EGFR)
- PMID: 25915403
- PMCID: PMC4411066
- DOI: 10.1371/journal.pone.0125793
Lipopolysaccharide (LPS)-Induced Biliary Epithelial Cell NRas Activation Requires Epidermal Growth Factor Receptor (EGFR)
Abstract
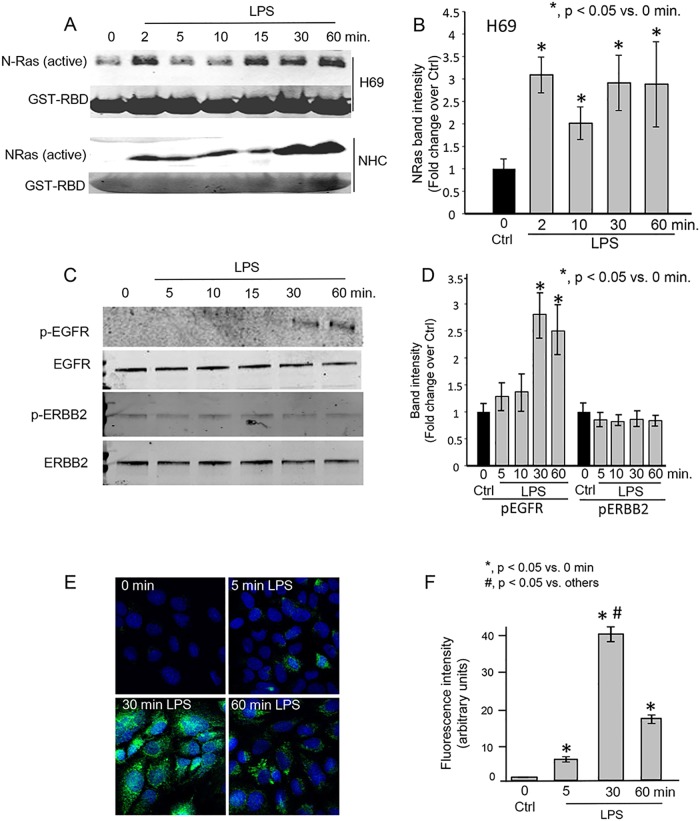
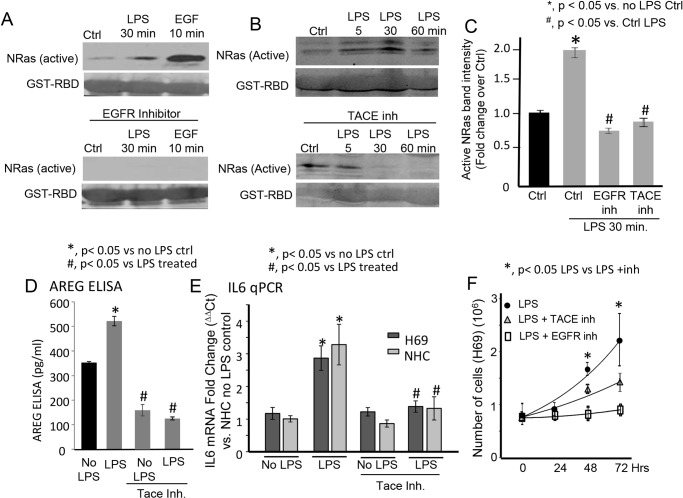
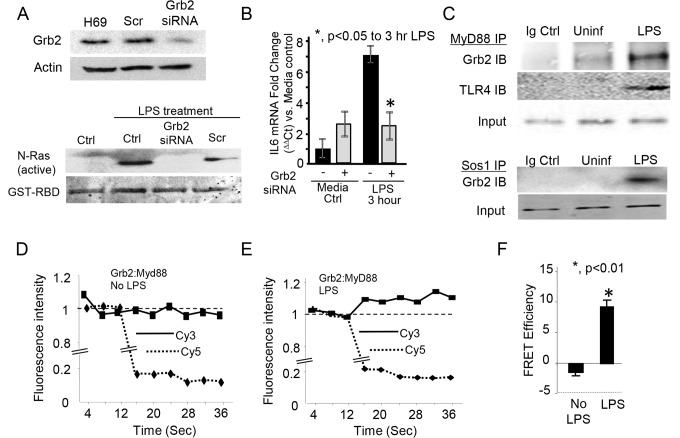
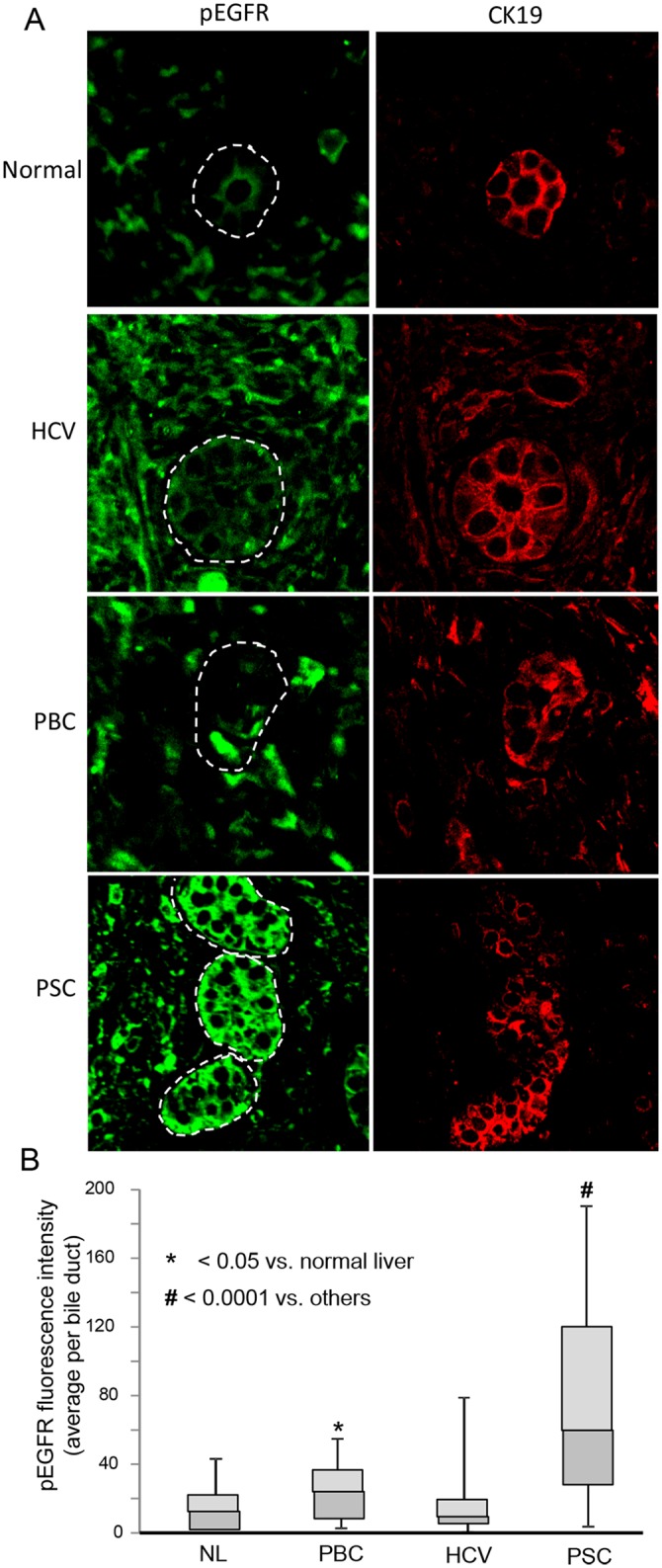
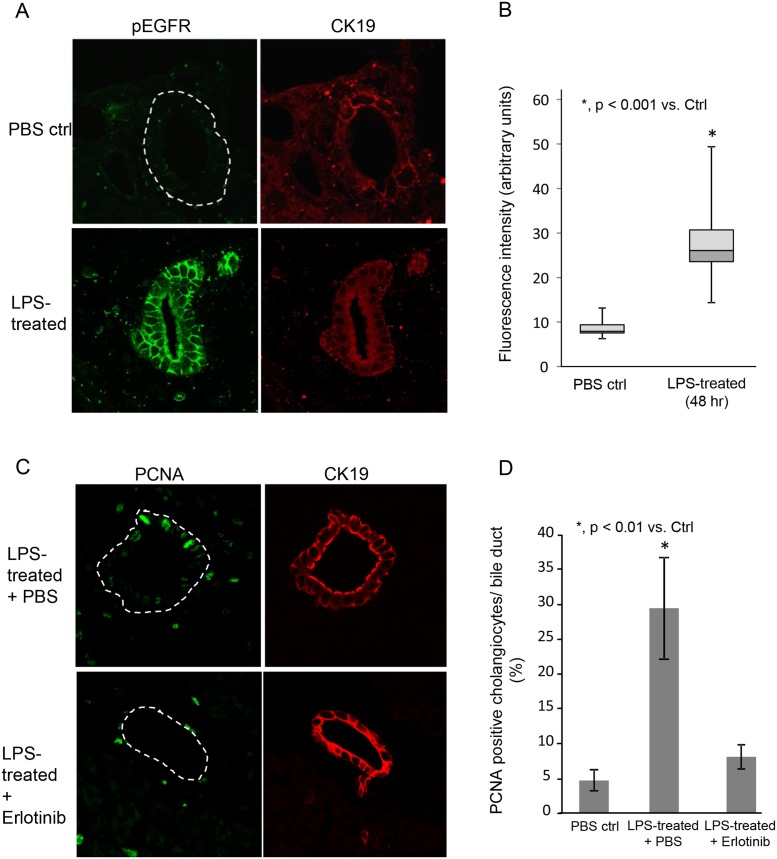
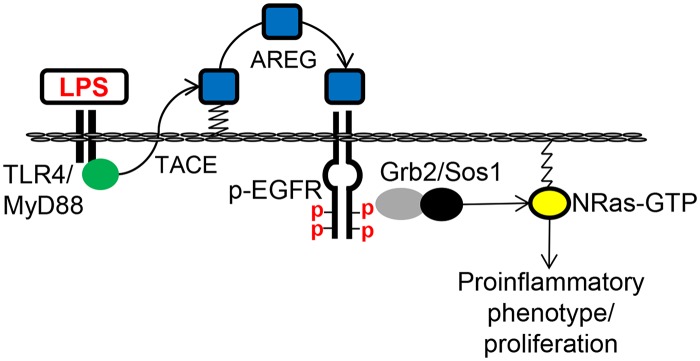
Cholangiocytes (biliary epithelial cells) actively participate in microbe-induced proinflammatory responses in the liver and contribute to inflammatory and infectious cholangiopathies. We previously demonstrated that cholangiocyte TLR-dependent NRas activation contributes to proinflammatory/ proliferative responses. We test the hypothesis that LPS-induced activation of NRas requires the EGFR. SV40-transformed human cholangiocytes (H69 cells), or low passage normal human cholangiocytes (NHC), were treated with LPS in the presence or absence of EGFR or ADAM metallopeptidase domain 17 (TACE) inhibitors. Ras activation assays, quantitative RT-PCR, and proliferation assays were performed in cells cultured with or without inhibitors or an siRNA to Grb2. Immunofluorescence for phospho-EGFR was performed on LPS-treated mouse samples and specimens from patients with primary sclerosing cholangitis, primary biliary cirrhosis, hepatitis C, and normal livers. LPS-treatment induced an association between the TLR/MyD88 and EGFR/Grb2 signaling apparatus, NRas activation, and EGFR phosphorylation. NRas activation was sensitive to EGFR and TACE inhibitors and correlated with EGFR phosphorylation. The TACE inhibitor and Grb2 depletion prevented LPS-induced IL6 expression (p<0.05) and proliferation (p<0.01). Additionally, cholangiocytes from LPS-treated mouse livers and human primary sclerosing cholangitis (PSC) livers exhibited increased phospho-EGFR (p<0.01). Moreover, LPS-induced mouse cholangiocyte proliferation was inhibited by concurrent treatment with the EGFR inhibitor, Erlotinib. Our results suggest that EGFR is essential for LPS-induced, TLR4/MyD88-mediated NRas activation and induction of a robust proinflammatory cholangiocyte response. These findings have implications not only for revealing the signaling potential of TLRs, but also implicate EGFR as an integral component of cholangiocyte TLR-induced proinflammatory processes.
Conflict of interest statement
Figures
References
-
- Harada K, Shimoda S, Sato Y, Isse K, Ikeda H, Nakanuma Y. Periductal interleukin-17 production in association with biliary innate immunity contributes to the pathogenesis of cholangiopathy in primary biliary cirrhosis. Clin Exp Immunol. 2009;157: 261–270. 10.1111/j.1365-2249.2009.03947.x - DOI - PMC - PubMed
-
- Karrar A, Broome U, Sodergren T, Jaksch M, Bergquist A, Bjornstedt M, et al. Biliary epithelial cell antibodies link adaptive and innate immune responses in primary sclerosing cholangitis. Gastroenterology. 2007;132: 1504–1514. - PubMed
-
- Mimura Y, Sakisaka S, Harada M, Sata M, Tanikawa K. Role of hepatocytes in direct clearance of lipopolysaccharide in rats. Gastroenterology. 1995;109: 1969–1976. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
