

HIV-1 and interferons: who's interfering with whom?
- PMID: 25915633
- PMCID: PMC7768976
- DOI: 10.1038/nrmicro3449
HIV-1 and interferons: who's interfering with whom?
Abstract
The ability of interferons (IFNs) to inhibit HIV-1 replication in cell culture models has long been recognized, and the therapeutic administration of IFNα to HIV-1-infected patients who are not receiving antiretroviral therapy produces a clear but transient decrease in plasma viral load. Conversely, studies of chronic HIV-1 infection in humans and SIV-infected animal models of AIDS show positive correlations between elevated plasma levels of IFNs, increased expression of IFN-stimulated genes (ISGs), biomarkers of inflammation and disease progression. In this Review, we discuss the evidence that IFNs can control HIV-1 replication in vivo and debate the controversial role of IFNs in promoting the pathological sequelae of chronic HIV-1 infection.
Conflict of interest statement
Competing interests statement
The authors declare no competing interests.
Figures

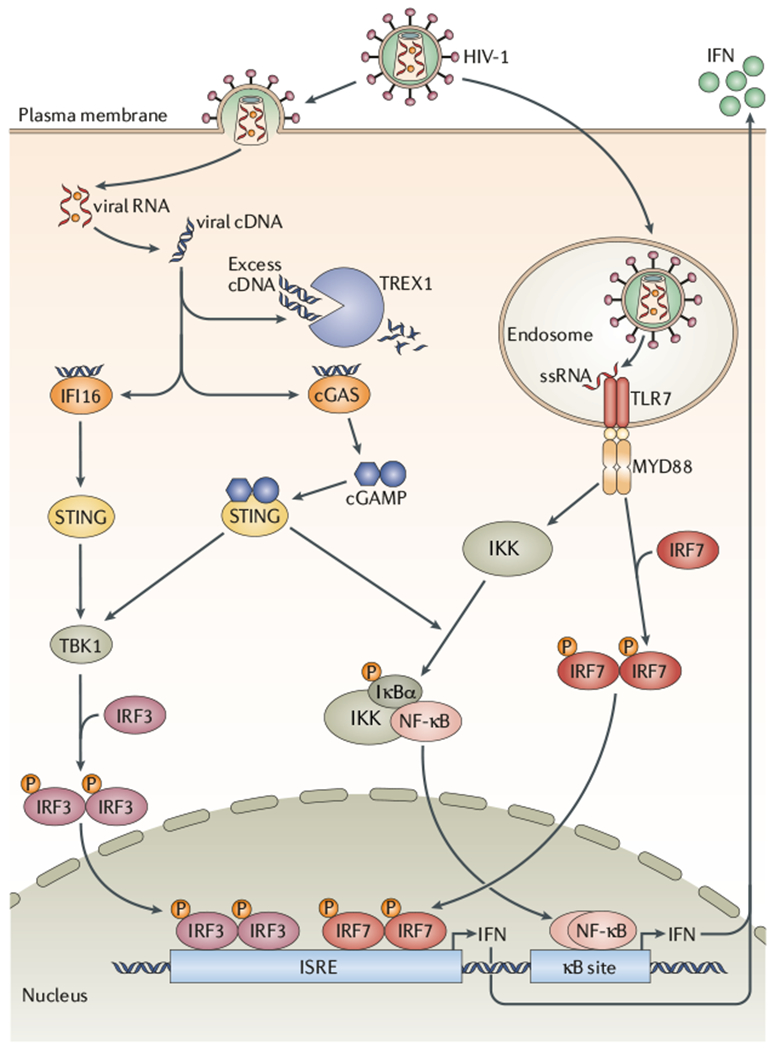


References
-
- Takeuchi O & Akira S Pattern recognition receptors and inflammation. Cell 140, 805–820 (2010). - PubMed
-
- Rotger M et al. Genome-wide mRNA expression correlates of viral control in CD4+ T-cells from HIV-1-infected individuals. PLoS Pathog. 6, e1000781 (2010). - PMC - PubMed
-
This study characterizes the gene expression profile abnormalities of the CD4+ T cells of patients with HIV-1 infection according to plasma viral load.
-
- Hyrcza MD et al. Distinct transcriptional profiles in ex vivo CD4+ and CD8+ T cells are established early in human immunodeficiency virus type 1 infection and are characterized by a chronic interferon response as well as extensive transcriptional changes in CD8+ T cells. J. Virol. 81, 3477–3486 (2007). - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
