

A statistical method for single sample analysis of HumanMethylation450 array data: genome-wide methylation analysis of patients with imprinting disorders
- PMID: 25918558
- PMCID: PMC4410592
- DOI: 10.1186/s13148-015-0081-5
A statistical method for single sample analysis of HumanMethylation450 array data: genome-wide methylation analysis of patients with imprinting disorders
Abstract
Background: The Illumina Infinium HumanMethylation450 BeadChip is an array-based technology for analysing DNA methylation at approximately 475,000 differentially methylated cytosines across the human genome. Hitherto, the array has been used for case-control studies, where sample numbers can be sufficient to yield statistically robust data on a genome-wide basis. We recently reported an informatic pipeline capable of yielding statistically and biologically significant results using only five cases, which expanded the use of this technology to rare disease studies. However, the clinical application of these technologies requires the ability to perform robust analysis of individual patients.
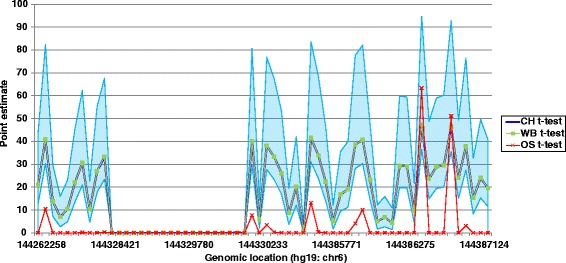
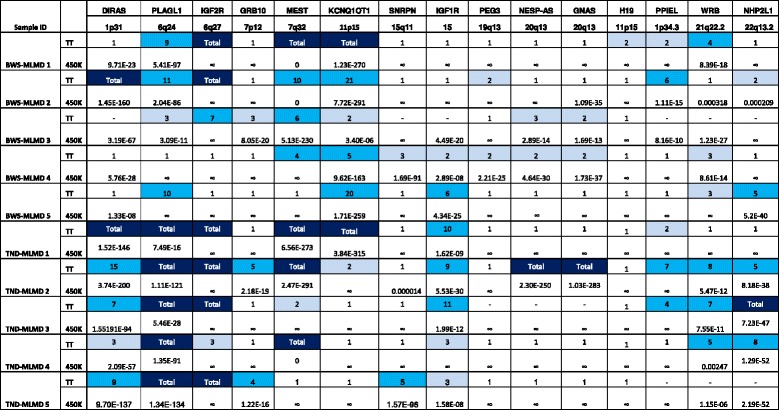
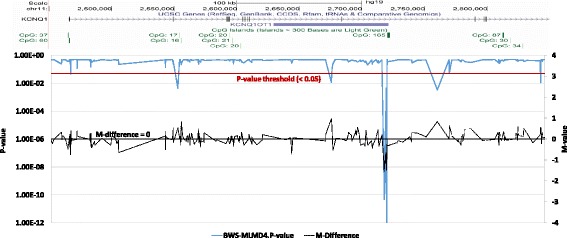
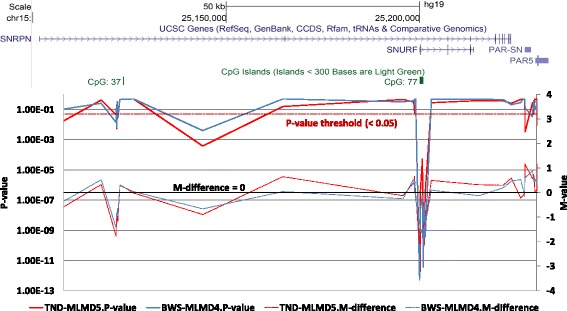
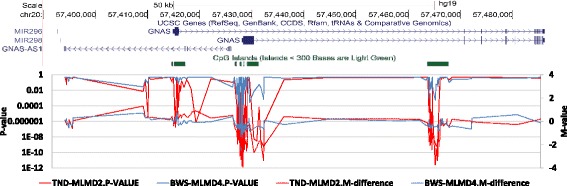
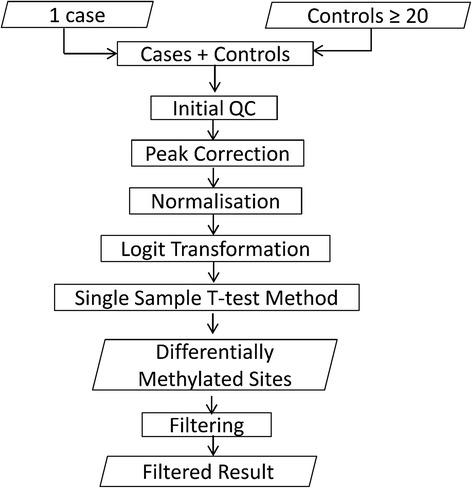
Results: Here we report a novel informatic approach for methylation array analysis of single samples, using the Crawford-Howell t-test. We tested our approach on patients with ultra-rare imprinting disorders with aberrant DNA methylation at multiple locations across the genome, which was previously detected by targeted testing. However, array analysis outperformed targeted assays in three ways: it detected loci not normally analysed by targeted testing, detected methylation changes too subtle to detect by the targeted testing and reported broad and consistent methylation changes across genetic loci not captured by point testing.
Conclusions: This method has potential clinical utility for human disorders where DNA methylation change may be a biomarker of disease.
Keywords: Crawford-Howell t-test; Illumina HumanMethylation450 array; Methylation; Single case-control analysis.
Figures
References
-
- Shenker NS, Polidoro S, van Veldhoven K, Sacerdote C, Ricceri F, Birrell MA, et al. Epigenome-wide association study in the European Prospective Investigation into Cancer and Nutrition (EPIC-Turin) identifies novel genetic loci associated with smoking. Hum Mol Genet. 2013;22:843–51. doi: 10.1093/hmg/dds488. - DOI - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
