

Whole Exome Sequencing Reveals the Order of Genetic Changes during Malignant Transformation and Metastasis in a Single Patient with NF1-plexiform Neurofibroma
- PMID: 25925892
- PMCID: PMC4573781
- DOI: 10.1158/1078-0432.CCR-14-3049
Whole Exome Sequencing Reveals the Order of Genetic Changes during Malignant Transformation and Metastasis in a Single Patient with NF1-plexiform Neurofibroma
Abstract
Purpose: Malignant peripheral nerve sheath tumors (MPNST) occur at increased frequency in individuals with neurofibromatosis type 1 (NF1), where they likely arise from benign plexiform neurofibroma precursors. While previous studies have used a variety of discovery approaches to discover genes associated with MPNST pathogenesis, it is currently unclear what molecular events are associated with the evolution of MPNST from plexiform neurofibroma.
Experimental design: Whole-exome sequencing was performed on biopsy materials representing plexiform neurofibroma (n = 3), MPNST, and metastasis from a single individual with NF1 over a 14-year period. Additional validation cases were used to assess candidate genes involved in malignant progression, while a murine MPNST model was used for functional analysis.
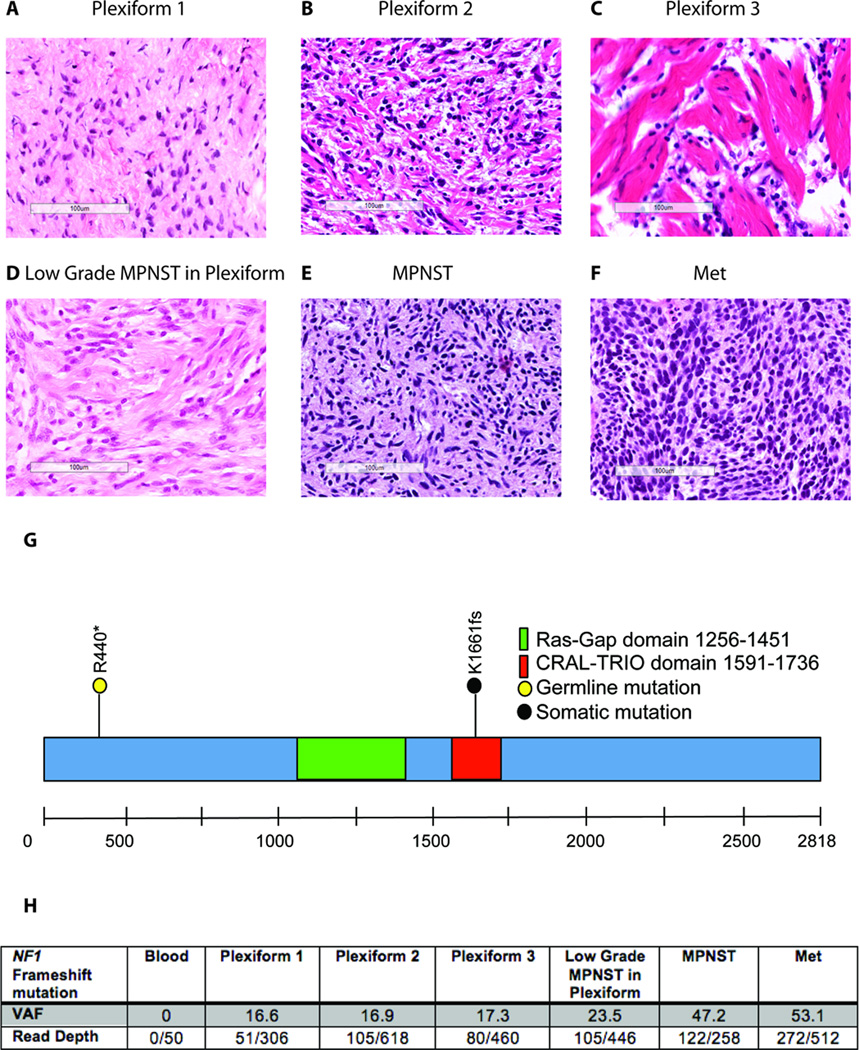
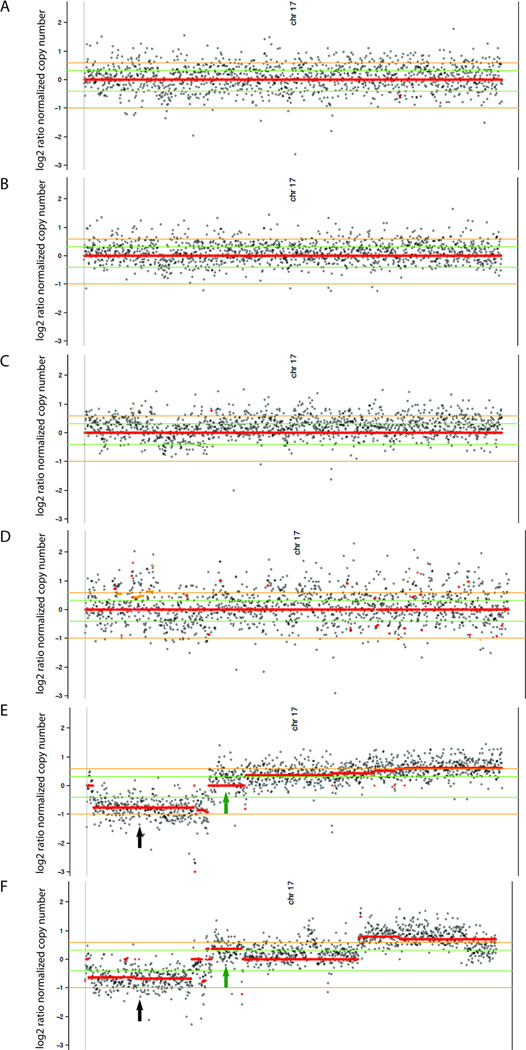
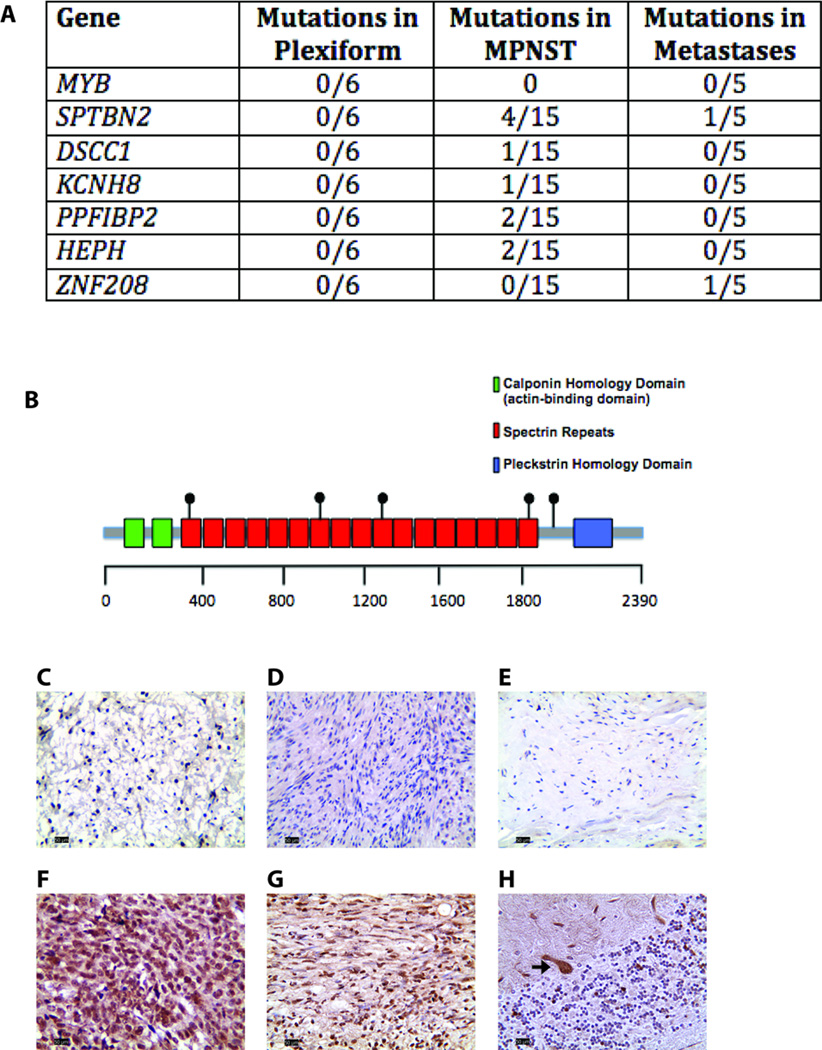
Results: There was an increasing proportion of cells with a somatic NF1 gene mutation as the tumors progressed from benign to malignant, suggesting a clonal process in MPNST development. Copy number variations, including loss of one copy of the TP53 gene, were identified in the primary tumor and the metastatic lesion, but not in benign precursor lesions. A limited number of genes with nonsynonymous somatic mutations (βIII-spectrin and ZNF208) were discovered, several of which were validated in additional primary and metastatic MPNST samples. Finally, increased βIII-spectrin expression was observed in the majority of MPNSTs, and shRNA-mediated knockdown reduced murine MPNST growth in vivo.
Conclusions: Collectively, the ability to track the molecular evolution of MPNST in a single individual with NF1 offers new insights into the sequence of genetic events important for disease pathogenesis and progression for future mechanistic study.
©2015 American Association for Cancer Research.
Conflict of interest statement
Figures

References
-
- Arun D, Gutmann DH. Recent advances in neurofibromatosis type 1. Current opinion in neurology. 2004;17:101–105. - PubMed
-
- Hirbe AC, Gutmann DH. Neurofibromatosis type 1: a multidisciplinary approach to care. The Lancet Neurology. 2014;13:834–843. - PubMed
-
- Upadhyaya M, Spurlock G, Monem B, Thomas N, Friedrich RE, Kluwe L, et al. Germline and somatic NF1 gene mutations in plexiform neurofibromas. Human mutation. 2008;29:E103–E111. - PubMed
-
- Rasmussen SA, Overman J, Thomson SA, Colman SD, Abernathy CR, Trimpert RE, et al. Chromosome 17 loss-of-heterozygosity studies in benign and malignant tumors in neurofibromatosis type 1. Genes, chromosomes & cancer. 2000;28:425–431. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
