

Clinical characterization of int22h1/int22h2-mediated Xq28 duplication/deletion: new cases and literature review
- PMID: 25927380
- PMCID: PMC4422130
- DOI: 10.1186/s12881-015-0157-2
Clinical characterization of int22h1/int22h2-mediated Xq28 duplication/deletion: new cases and literature review
Abstract
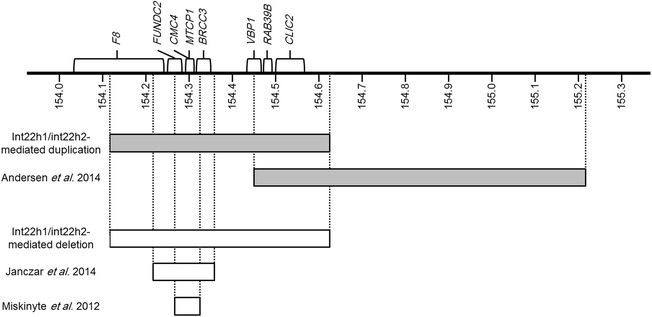
Background: Int22h1/int22h2-mediated Xq28 duplication syndrome is caused by ~0.5 Mb chromosomal duplications mediated by nonallelic homologous recombination between intron 22 homologous region 1 (int22h1) and 2 (int22h2), which, in addition to int22h3, are also responsible for inversions disrupting the F8 gene in hemophilia A. This syndrome has recently been described in 9 males with cognitive impairment, behavioral problems, and distinctive facial features; and 6 females with milder phenotypes. The reciprocal deletion was previously reported in a mother and daughter. It was suggested that this deletion may not have phenotypic effects in females because of skewed chromosome X inactivation, but may be embryonic lethal in males.
Methods: Array comparative genomic hybridization analyses were performed using oligonucleotide-based chromosomal microarray. Chromosome X inactivation studies were performed at the AR (androgen receptor) and FMR1 (fragile X mental retardation 1) loci.
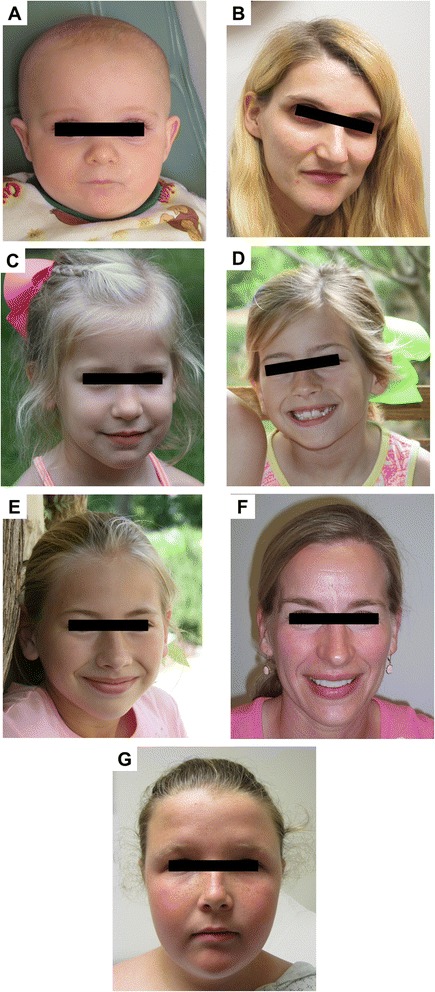
Results: We present here 5 males and 6 females with int22h1/int22h2-mediated Xq28 duplication syndrome. The males manifested cognitive impairment, behavioral problems, and distinctive facial features. Two of the six females manifested mild cognitive impairment. This duplication was maternally inherited, and skewed chromosome X inactivation was observed in the majority of females carrying the duplication. We also report the reciprocal deletion in a mother and daughter with overweight, but normal cognition. In addition, we present the first case of a prenatally diagnosed de novo int22h1/int22h2-mediated deletion in a healthy female infant. We reviewed individuals previously reported with similar or overlapping rearrangements and evaluated the potential roles of genes in the rearrangement region.
Conclusions: The similarity of clinical features among individuals with the int22h1/int22h2-mediated Xq28 duplication supports the notion that this duplication causes a recognizable syndrome that affects males with females exhibiting milder phenotypes. It is suggested that the observed cognitive impairment in this syndrome results from increased dosage of RAB39B gene located within the duplicated region. Increased dosage of CLIC2 may also contribute to the phenotype. The reciprocal deletion results in skewed chromosome X inactivation and no clinical phenotype in females. Review of overlapping deletions suggests that hemizygous loss of VBP1 may be the cause for the proposed male lethality associated with this deletion.
Figures
References
-
- Lisik MZ, Sieron AL. X-linked mental retardation. Med Sci Monit. 2008;14:221–9. - PubMed
-
- Froyen G, Van Esch H, Bauters M, Hollanders K, Frints SG, Vermeesch JR, et al. Detection of genomic copy number changes in patients with idiopathic mental retardation by High-Resolution X-Array-CGH: important role for increased gene dosage of XLMR genes. Hum Mut. 2007;28:1042–3. doi: 10.1002/humu.20564. - DOI - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
