

ContextMap 2: fast and accurate context-based RNA-seq mapping
- PMID: 25928589
- PMCID: PMC4411664
- DOI: 10.1186/s12859-015-0557-5
ContextMap 2: fast and accurate context-based RNA-seq mapping
Abstract
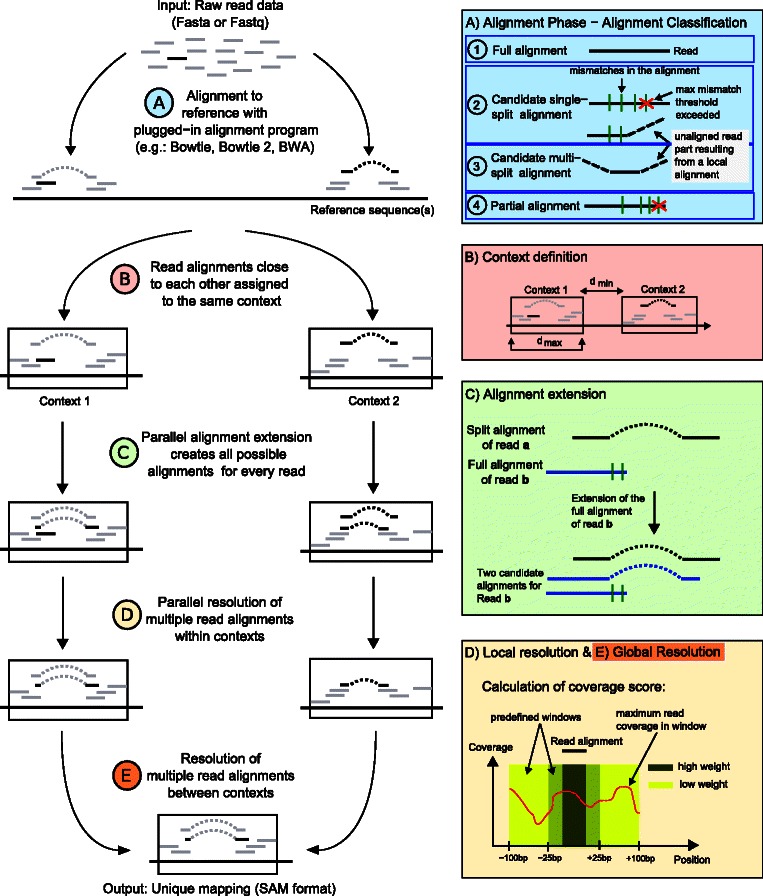
Background: Mapping of short sequencing reads is a crucial step in the analysis of RNA sequencing (RNA-seq) data. ContextMap is an RNA-seq mapping algorithm that uses a context-based approach to identify the best alignment for each read and allows parallel mapping against several reference genomes.
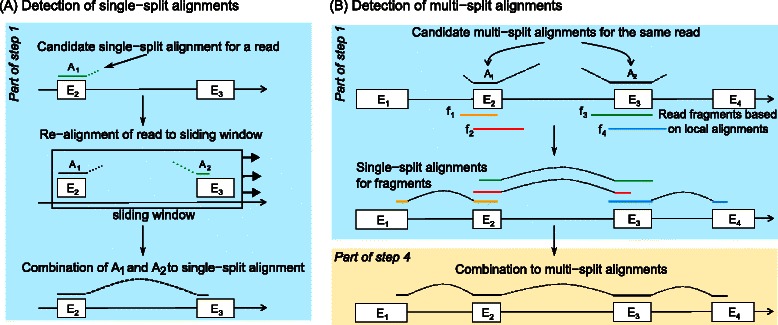
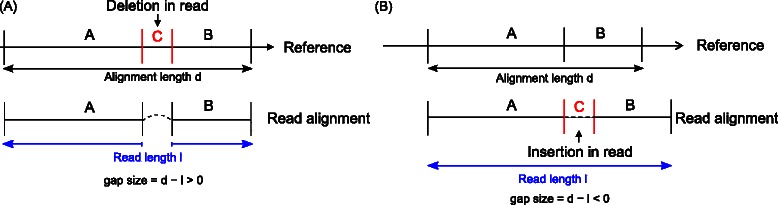
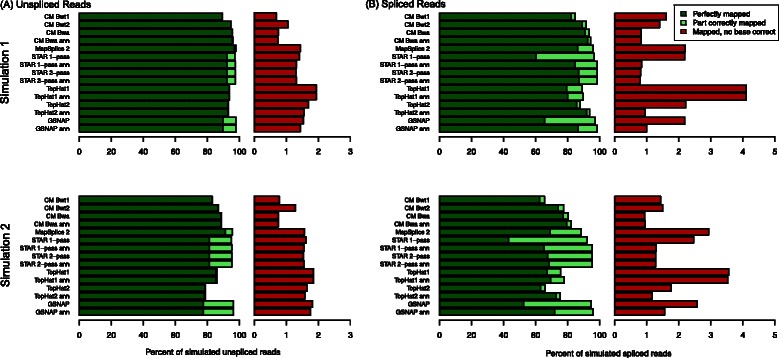
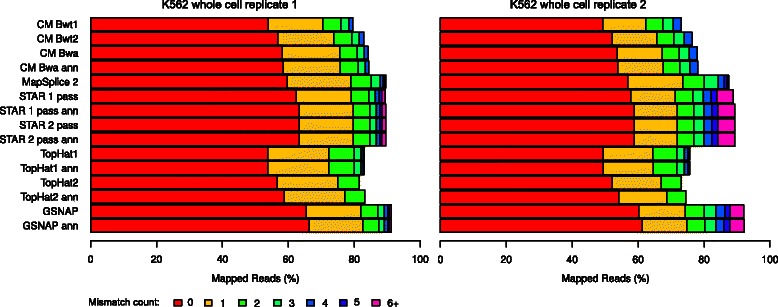
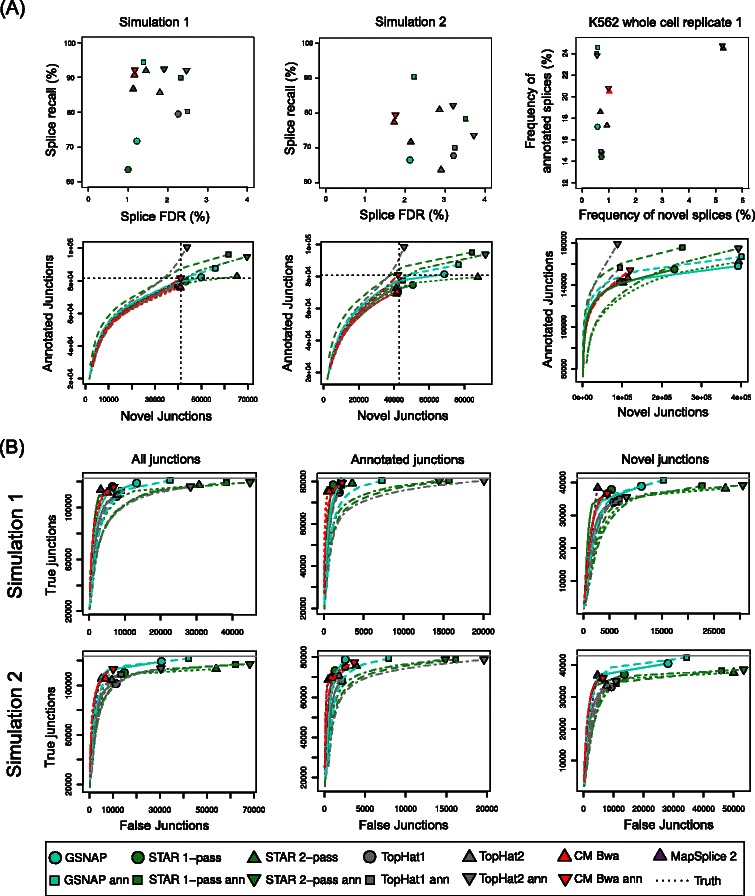
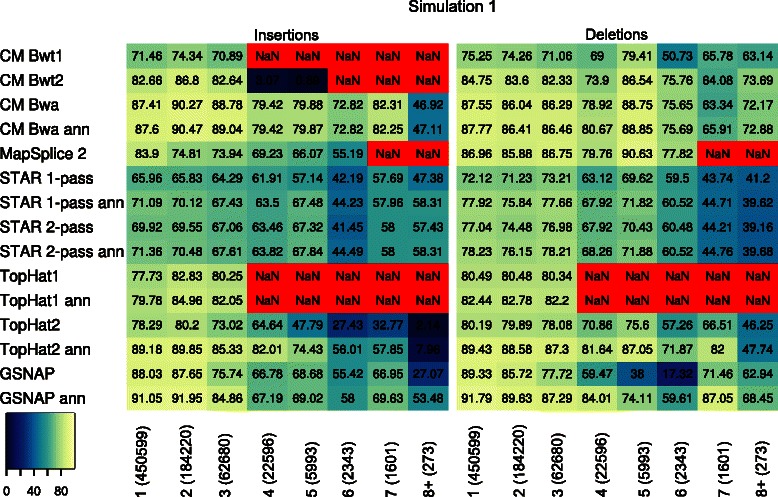
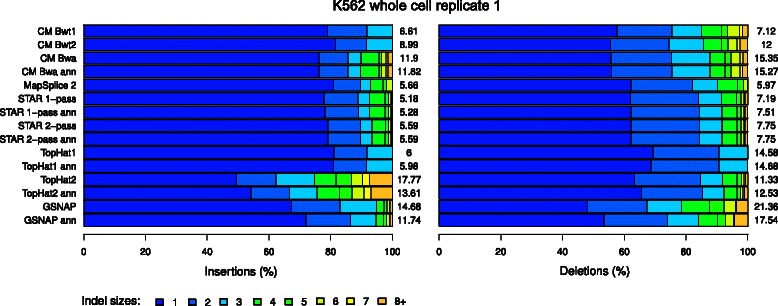
Results: In this article, we present ContextMap 2, a new and improved version of ContextMap. Its key novel features are: (i) a plug-in structure that allows easily integrating novel short read alignment programs with improved accuracy and runtime; (ii) context-based identification of insertions and deletions (indels); (iii) mapping of reads spanning an arbitrary number of exons and indels. ContextMap 2 using Bowtie, Bowtie 2 or BWA was evaluated on both simulated and real-life data from the recently published RGASP study.
Conclusions: We show that ContextMap 2 generally combines similar or higher recall compared to other state-of-the-art approaches with significantly higher precision in read placement and junction and indel prediction. Furthermore, runtime was significantly lower than for the best competing approaches. ContextMap 2 is freely available at http://www.bio.ifi.lmu.de/ContextMap .
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
