

Genomic prediction of breeding values using previously estimated SNP variances
- PMID: 25928875
- PMCID: PMC4176585
- DOI: 10.1186/s12711-014-0052-x
Genomic prediction of breeding values using previously estimated SNP variances
Abstract
Background: Genomic prediction requires estimation of variances of effects of single nucleotide polymorphisms (SNPs), which is computationally demanding, and uses these variances for prediction. We have developed models with separate estimation of SNP variances, which can be applied infrequently, and genomic prediction, which can be applied routinely.
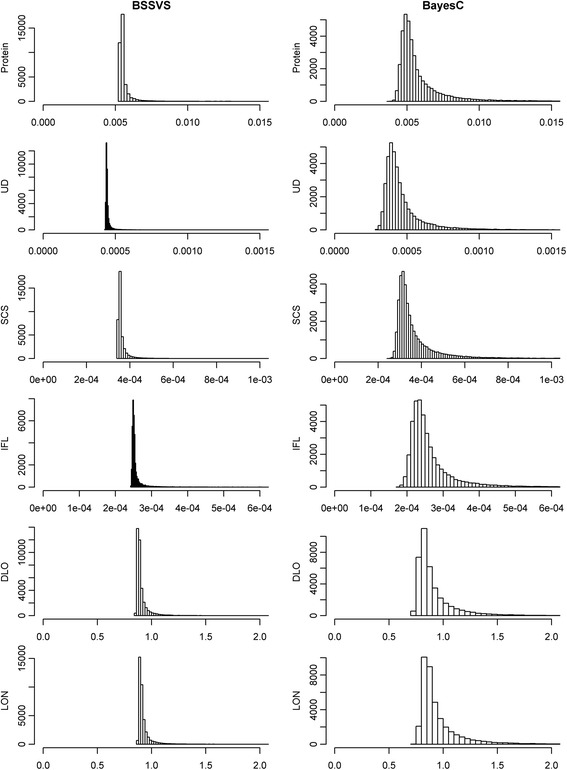
Methods: SNP variances were estimated with Bayes Stochastic Search Variable Selection (BSSVS) and BayesC. Genome-enhanced breeding values (GEBV) were estimated with RR-BLUP (ridge regression best linear unbiased prediction), using either variances obtained from BSSVS (BLUP-SSVS) or BayesC (BLUP-C), or assuming equal variances for each SNP. Datasets used to estimate SNP variances comprised (1) all animals, (2) 50% random animals (RAN50), (3) 50% best animals (TOP50), or (4) 50% worst animals (BOT50). Traits analysed were protein yield, udder depth, somatic cell score, interval between first and last insemination, direct longevity, and longevity including information from predictors.
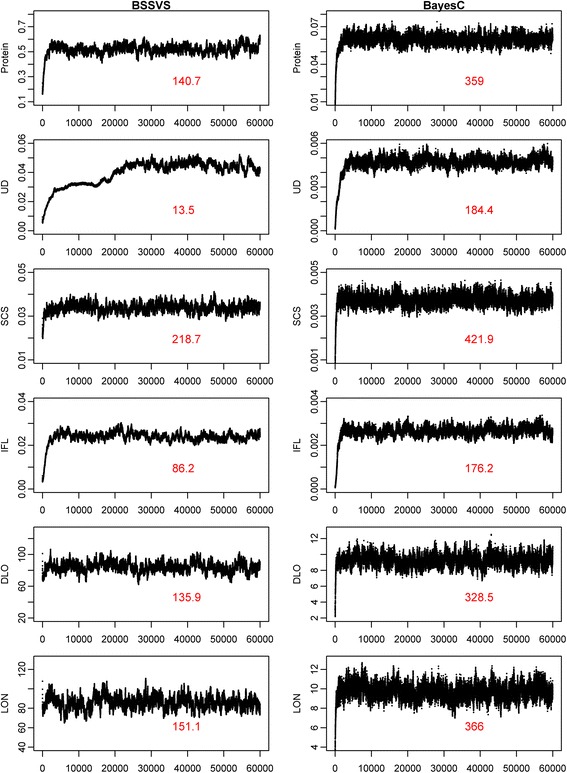
Results: BLUP-SSVS and BLUP-C yielded similar GEBV as the equivalent Bayesian models that simultaneously estimated SNP variances. Reliabilities of these GEBV were consistently higher than from RR-BLUP, although only significantly for direct longevity. Across scenarios that used data subsets to estimate GEBV, observed reliabilities were generally higher for TOP50 than for RAN50, and much higher than for BOT50. Reliabilities of TOP50 were higher because the training data contained more ancestors of selection candidates. Using estimated SNP variances based on random or non-random subsets of the data, while using all data to estimate GEBV, did not affect reliabilities of the BLUP models. A convergence criterion of 10(-8) instead of 10(-10) for BLUP models yielded similar GEBV, while the required number of iterations decreased by 71 to 90%. Including a separate polygenic effect consistently improved reliabilities of the GEBV, but also substantially increased the required number of iterations to reach convergence with RR-BLUP. SNP variances converged faster for BayesC than for BSSVS.
Conclusions: Combining Bayesian variable selection models to re-estimate SNP variances and BLUP models that use those SNP variances, yields GEBV that are similar to those from full Bayesian models. Moreover, these combined models yield predictions with higher reliability and less bias than the commonly used RR-BLUP model.
Figures
References
-
- Daetwyler HD, Capitan A, Pausch H, Stothard P, Van Binsbergen R, Brøndum RF, Liao X, Djari A, Rodriguez S, Grohs C, Jung S, Esquerré D, Bouchez O, Rossignol MN, Klopp C, Rocha D, Fritz S, Eggen A, Bowman P, Coote D, Chamberlain A, Vantassell CP, Hulsegge I, Goddard ME, Guldbrandtsen B, Lund MS, Veerkamp RF, Boichard DA, Fries R, Hayes BJ. Whole-genome sequencing of 234 bulls facilitates mapping of monogenic and complex traits in cattle. Nat Rev Genet. 2014;46:858–865. doi: 10.1038/ng.3034. - DOI - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
