

TLR2 and TLR9 contribute to alcohol-mediated liver injury through induction of CXCL1 and neutrophil infiltration
- PMID: 25930080
- PMCID: PMC4491507
- DOI: 10.1152/ajpgi.00031.2015
TLR2 and TLR9 contribute to alcohol-mediated liver injury through induction of CXCL1 and neutrophil infiltration
Abstract
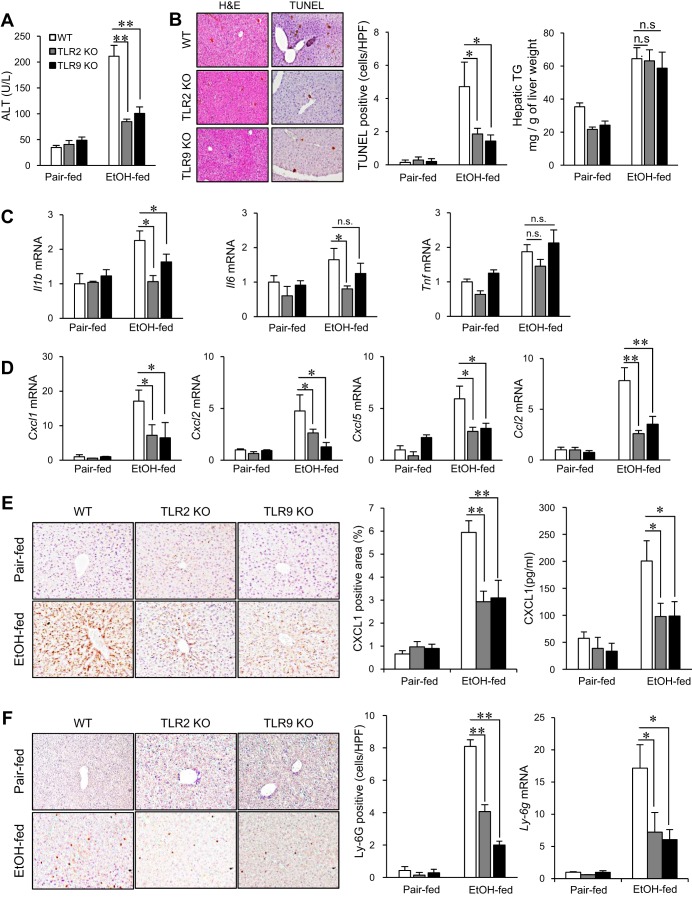
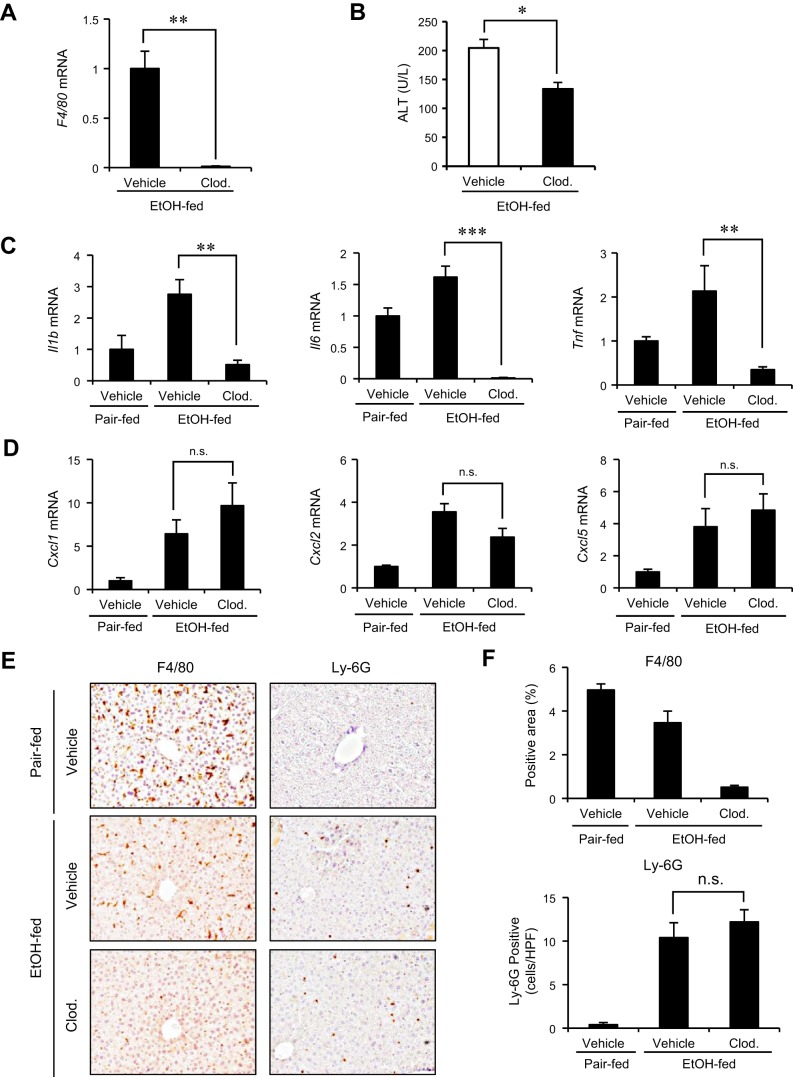
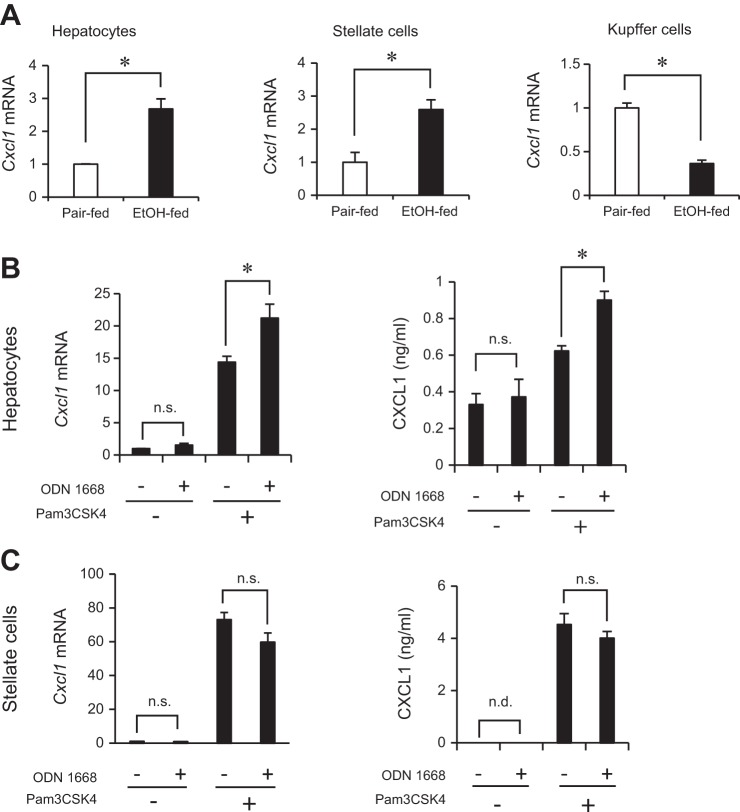
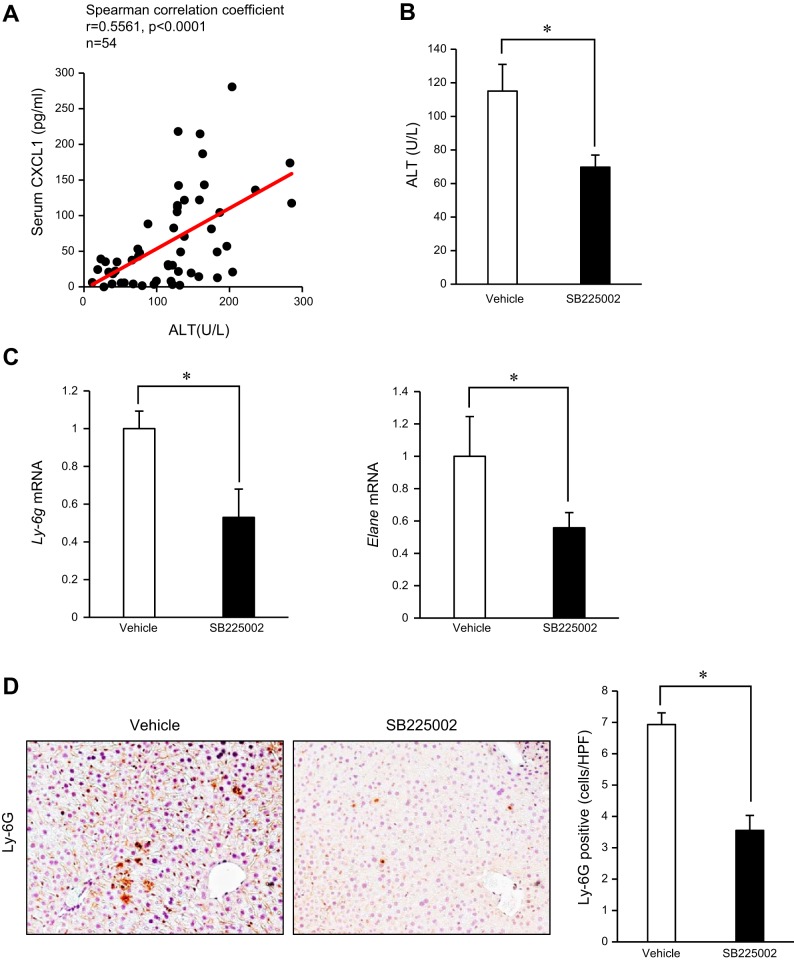
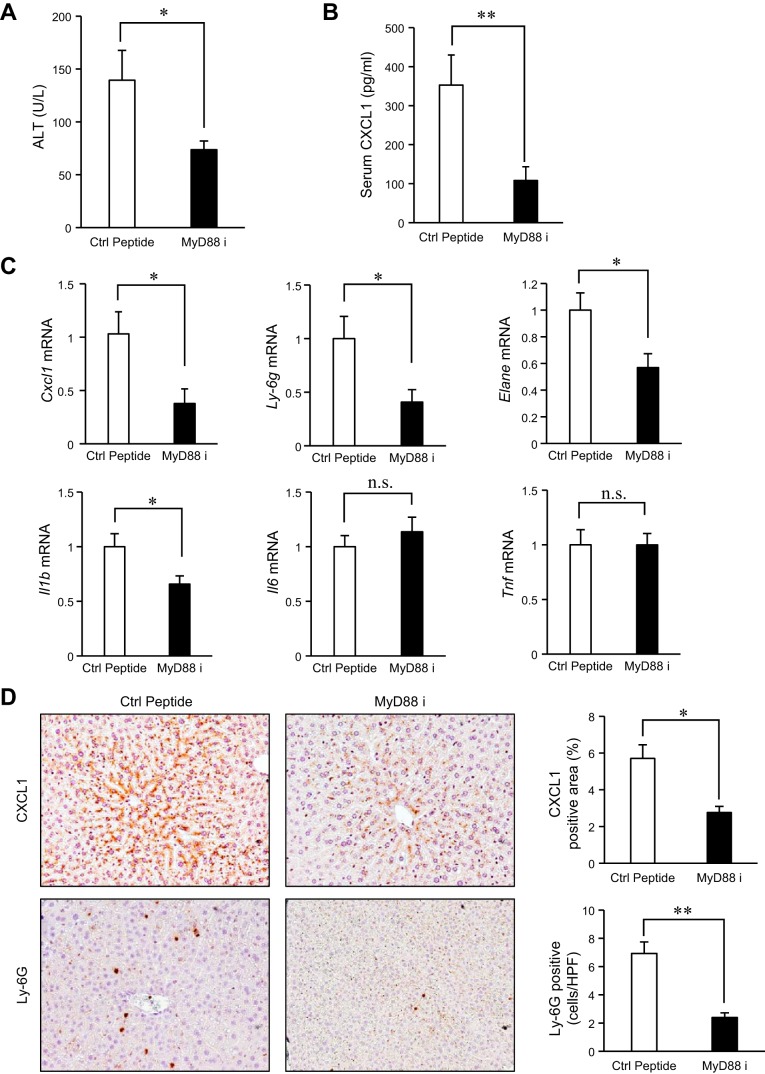
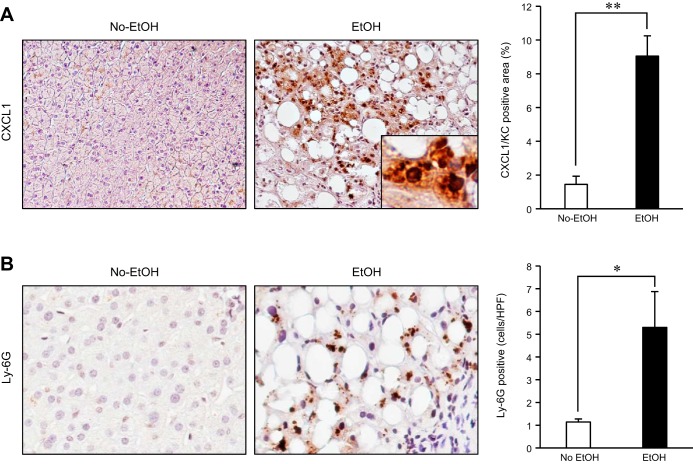
Although previous studies reported the involvement of the TLR4-TRIF pathway in alcohol-induced liver injury, the role of TLR2 and TLR9 signaling in alcohol-mediated neutrophil infiltration and liver injury has not been elucidated. Since alcohol binge drinking is recognized to induce more severe form of alcohol liver disease, we used a chronic-binge ethanol-feeding model as a mouse model for early stage of alcoholic hepatitis. Whereas a chronic-binge ethanol feeding induced alcohol-mediated liver injury in wild-type mice, TLR2- and TLR9-deficient mice showed reduced liver injury. Induction of neutrophil-recruiting chemokines, including Cxcl1, Cxcl2, and Cxcl5, and hepatic neutrophil infiltration were increased in wild-type mice, but not in TLR2- and TLR9-deficient mice. In vivo depletion of Kupffer cells (KCs) by liposomal clodronate reduced liver injury and the expression of Il1b, but not Cxcl1, Cxcl2, and Cxcl5, suggesting that KCs are partly associated with liver injury, but not neutrophil recruitment, in a chronic-binge ethanol-feeding model. Notably, hepatocytes and hepatic stellate cells (HSCs) produce high amounts of CXCL1 in ethanol-treated mice. The treatment with TLR2 and TLR9 ligands synergistically upregulated CXCL1 expression in hepatocytes. Moreover, the inhibitors for CXCR2, a receptor for CXCL1, and MyD88 suppressed neutrophil infiltration and liver injury induced by chronic-binge ethanol treatment. Consistent with the above findings, hepatic CXCL1 expression was highly upregulated in patients with alcoholic hepatitis. In a chronic-binge ethanol-feeding model, the TLR2 and TLR9-dependent MyD88-dependent pathway mediates CXCL1 production in hepatocytes and HSCs; the CXCL1 then promotes neutrophil infiltration into the liver via CXCR2, resulting in the development of alcohol-mediated liver injury.
Keywords: AH; ALD; MyD88; binge ethanol feeding; chemokine.
Copyright © 2015 the American Physiological Society.
Figures
References
-
- Adachi Y, Moore LE, Bradford BU, Gao W, Thurman RG. Antibiotics prevent liver injury in rats following long-term exposure to ethanol. Gastroenterology 108: 218–224, 1995. - PubMed
-
- Akira S, Hemmi H. Recognition of pathogen-associated molecular patterns by TLR family. Immunol Lett 85: 85–95, 2003. - PubMed
-
- Alho H, Sillanaukee P, Kalela A, Jaakkola O, Laine S, Nikkari ST. Alcohol misuse increases serum antibodies to oxidized LDL and C-reactive protein. Alcohol Alcohol 39: 312–315, 2004. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases