

Transgenic animal models for study of the pathogenesis of Huntington's disease and therapy
- PMID: 25931812
- PMCID: PMC4404937
- DOI: 10.2147/DDDT.S58470
Transgenic animal models for study of the pathogenesis of Huntington's disease and therapy
Abstract
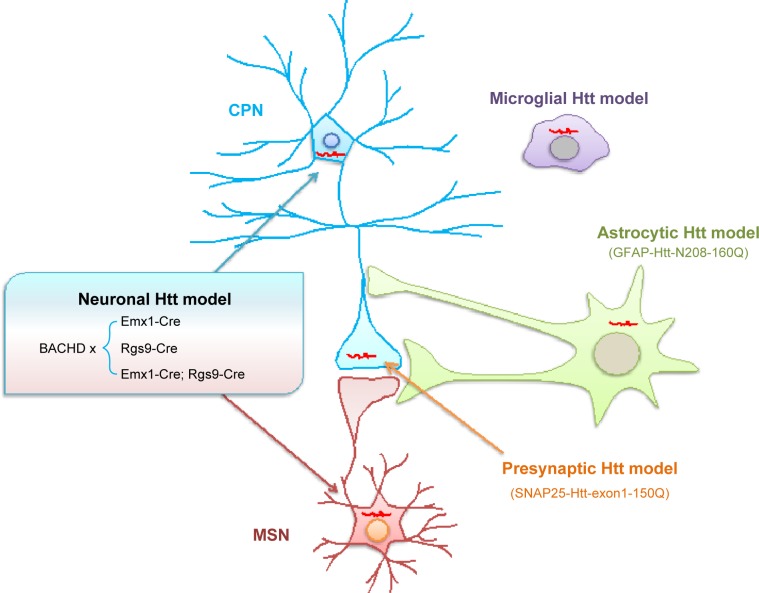
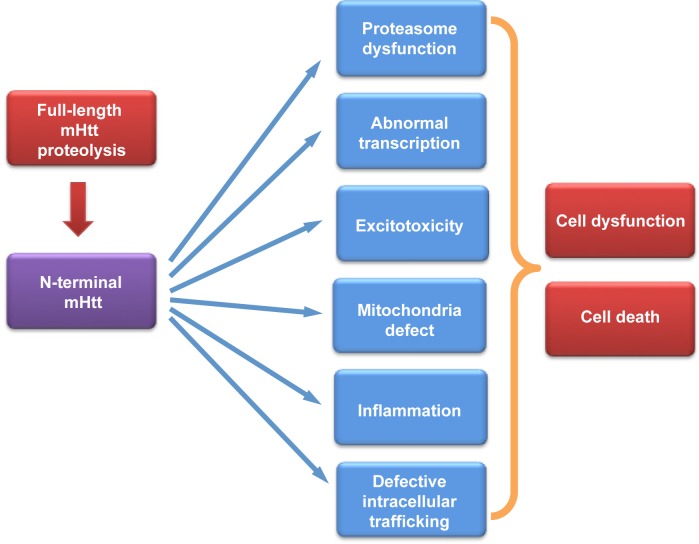
Huntington's disease (HD) is caused by a genetic mutation that results in polyglutamine expansion in the N-terminal regions of huntingtin. As a result, this polyQ expansion leads to the misfolding and aggregation of mutant huntingtin as well as age-dependent neurodegeneration. The genetic mutation in HD allows for generating a variety of animal models that express different forms of mutant huntingtin and show differential pathology. Studies of these animal models have provided an important insight into the pathogenesis of HD. Mouse models of HD include transgenic mice, which express N-terminal or full-length mutant huntingtin ubiquitously or selectively in different cell types, and knock-in mice that express full-length mutant Htt at the endogenous level. Large animals, such as pig, sheep, and monkeys, have also been used to generate animal HD models. This review focuses on the different features of commonly used transgenic HD mouse models as well as transgenic large animal models of HD, and also discusses how to use them to identify potential therapeutics. Since HD shares many pathological features with other neurodegenerative diseases, identification of therapies for HD would also help to develop effective treatment for different neurodegenerative diseases that are also caused by protein misfolding and occur in an age-dependent manner.
Keywords: Huntington’s disease; pathogenesis; therapy; transgenic animal models.
Figures
Similar articles
-
Progressive phenotype and nuclear accumulation of an amino-terminal cleavage fragment in a transgenic mouse model with inducible expression of full-length mutant huntingtin.Neurobiol Dis. 2006 Feb;21(2):381-91. doi: 10.1016/j.nbd.2005.07.014. Epub 2005 Sep 16. Neurobiol Dis. 2006. PMID: 16150600
-
Formation of polyglutamine inclusions in a wide range of non-CNS tissues in the HdhQ150 knock-in mouse model of Huntington's disease.PLoS One. 2009 Nov 30;4(11):e8025. doi: 10.1371/journal.pone.0008025. PLoS One. 2009. PMID: 19956633 Free PMC article.
-
Green tea (-)-epigallocatechin-gallate modulates early events in huntingtin misfolding and reduces toxicity in Huntington's disease models.Hum Mol Genet. 2006 Sep 15;15(18):2743-51. doi: 10.1093/hmg/ddl210. Epub 2006 Aug 7. Hum Mol Genet. 2006. PMID: 16893904
-
Large animal models for Huntington's disease research.Zool Res. 2024 Mar 18;45(2):275-283. doi: 10.24272/j.issn.2095-8137.2023.199. Zool Res. 2024. PMID: 38485497 Free PMC article. Review.
-
Animal models of Huntington's disease: implications in uncovering pathogenic mechanisms and developing therapies.Acta Pharmacol Sin. 2006 Oct;27(10):1287-302. doi: 10.1111/j.1745-7254.2006.00410.x. Acta Pharmacol Sin. 2006. PMID: 17007735 Review.
Cited by
-
Huntingtin N17 domain is a reactive oxygen species sensor regulating huntingtin phosphorylation and localization.Hum Mol Genet. 2016 Sep 15;25(18):3937-3945. doi: 10.1093/hmg/ddw234. Epub 2016 Jul 27. Hum Mol Genet. 2016. PMID: 27466181 Free PMC article.
-
In vitro modeling of the neurovascular unit: advances in the field.Fluids Barriers CNS. 2020 Mar 16;17(1):22. doi: 10.1186/s12987-020-00183-7. Fluids Barriers CNS. 2020. PMID: 32178700 Free PMC article. Review.
-
The Potential Regulatory Mechanisms of miR-196a in Huntington's Disease through Bioinformatic Analyses.PLoS One. 2015 Sep 16;10(9):e0137637. doi: 10.1371/journal.pone.0137637. eCollection 2015. PLoS One. 2015. PMID: 26376480 Free PMC article.
-
Bridging the gap: large animal models in neurodegenerative research.Mamm Genome. 2017 Aug;28(7-8):324-337. doi: 10.1007/s00335-017-9687-6. Epub 2017 Apr 4. Mamm Genome. 2017. PMID: 28378063 Free PMC article. Review.
-
TSPO imaging in animal models of brain diseases.Eur J Nucl Med Mol Imaging. 2021 Dec;49(1):77-109. doi: 10.1007/s00259-021-05379-z. Epub 2021 Jul 10. Eur J Nucl Med Mol Imaging. 2021. PMID: 34245328 Free PMC article. Review.
References
-
- Macdonald ME, Ambrose CM, Duyao MP, et al. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntingtons disease chromosomes. Cell. 1993;72(6):971–983. - PubMed
-
- Duyao MP, Ambrose CM, Myers RH, et al. Trinucleotide repeat length – instability and age-of-onset in Huntingtons disease. Am J Hum Genet. 1993;53(3):1152. - PubMed
-
- Stine OC, Pleasant N, Franz ML, Abbott MH, Folstein SE, Ross CA. Correlation between the onset age of Huntingtons-disease and length of the trinucleotide repeat in It-15. Hum Mol Genet. 1993;2(10):1547–1549. - PubMed
-
- Harper PS. Huntington’s Disease. 2nd ed. London, UK: WB Saunders; 1996.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
