

Emerging mechanisms of molecular pathology in ALS
- PMID: 25932674
- PMCID: PMC4463186
- DOI: 10.1172/JCI71601
Emerging mechanisms of molecular pathology in ALS
Erratum in
-
Emerging mechanisms of molecular pathology in ALS.J Clin Invest. 2015 Jun;125(6):2548. doi: 10.1172/JCI82693. Epub 2015 Jun 1. J Clin Invest. 2015. PMID: 26030230 Free PMC article. No abstract available.
Abstract
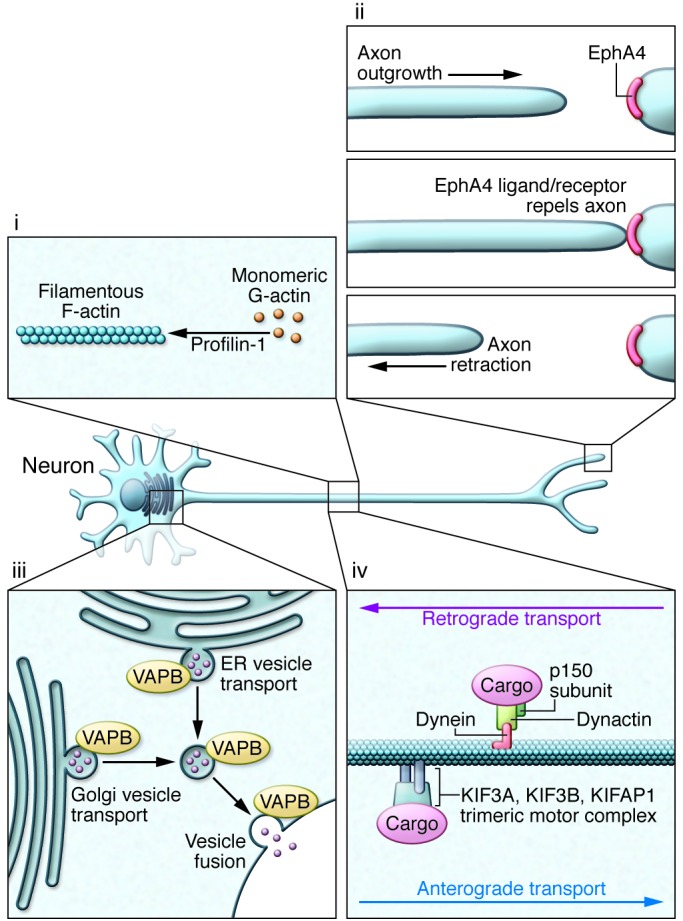
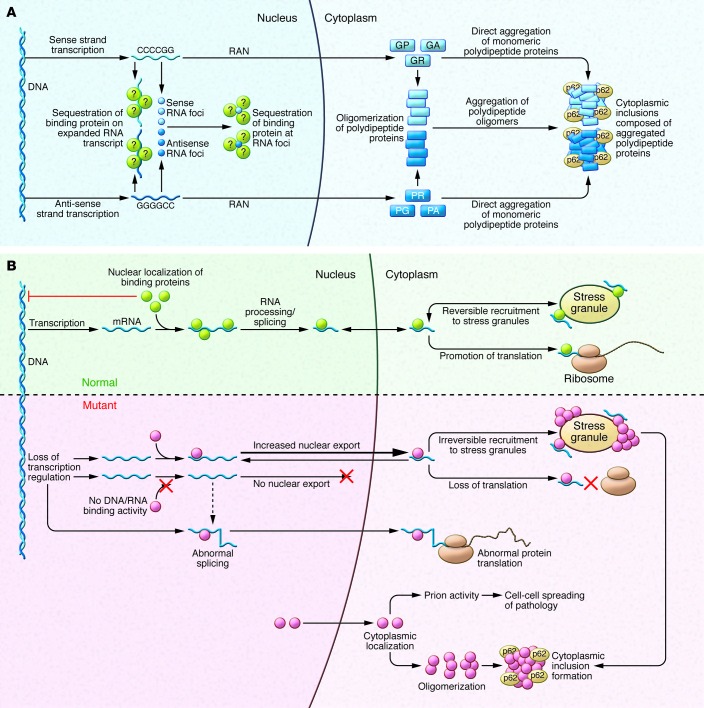
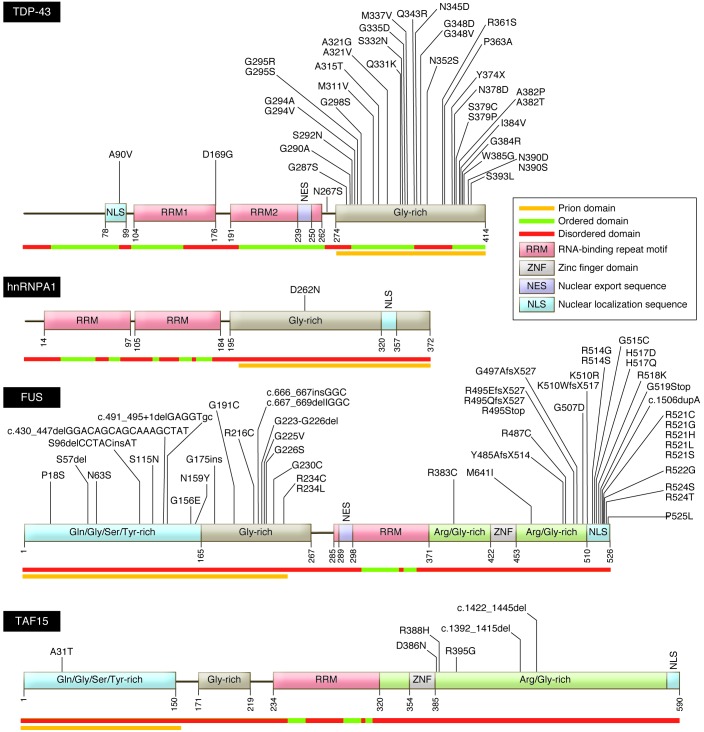
Amyotrophic lateral sclerosis (ALS) is a devastating degenerative disease characterized by progressive loss of motor neurons in the motor cortex, brainstem, and spinal cord. Although defined as a motor disorder, ALS can arise concurrently with frontotemporal lobal dementia (FTLD). ALS begins focally but disseminates to cause paralysis and death. About 10% of ALS cases are caused by gene mutations, and more than 40 ALS-associated genes have been identified. While important questions about the biology of this disease remain unanswered, investigations of ALS genes have delineated pathogenic roles for (a) perturbations in protein stability and degradation, (b) altered homeostasis of critical RNA- and DNA-binding proteins, (c) impaired cytoskeleton function, and (d) non-neuronal cells as modifiers of the ALS phenotype. The rapidity of progress in ALS genetics and the subsequent acquisition of insights into the molecular biology of these genes provide grounds for optimism that meaningful therapies for ALS are attainable.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
