

Plasminogen activator inhibitor-1 in cigarette smoke exposure and influenza A virus infection-induced lung injury
- PMID: 25932922
- PMCID: PMC4416821
- DOI: 10.1371/journal.pone.0123187
Plasminogen activator inhibitor-1 in cigarette smoke exposure and influenza A virus infection-induced lung injury
Abstract
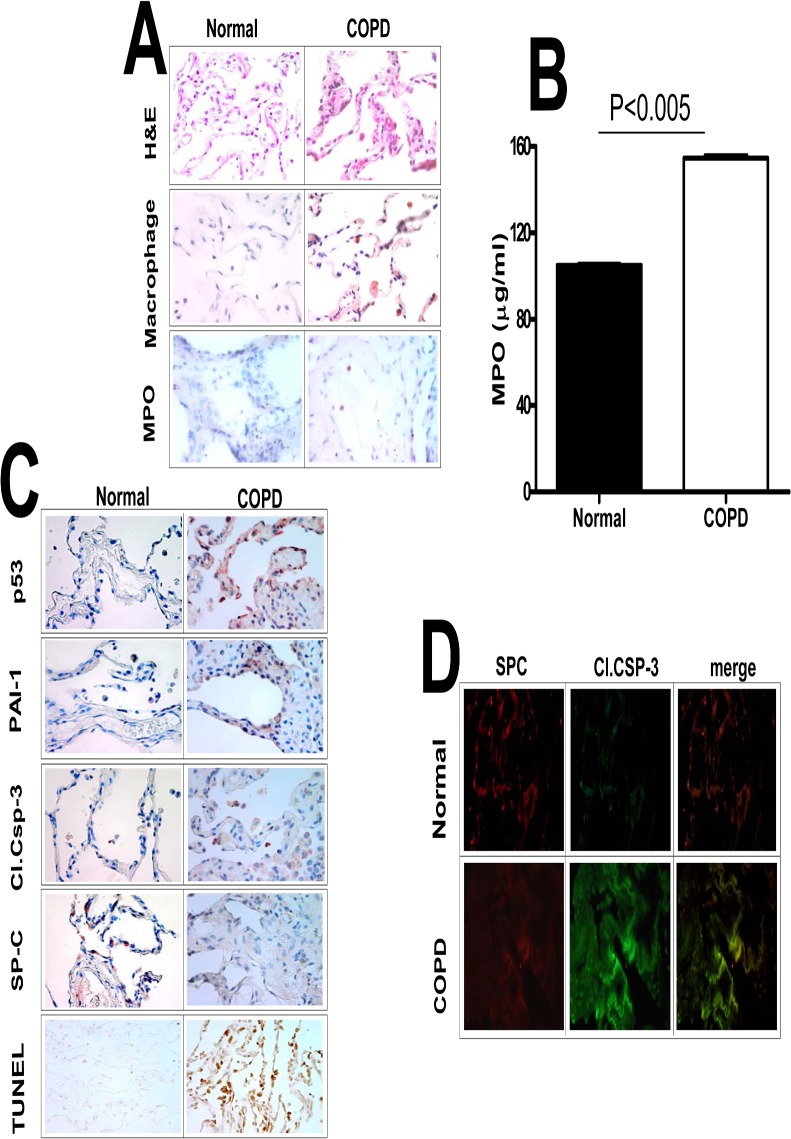
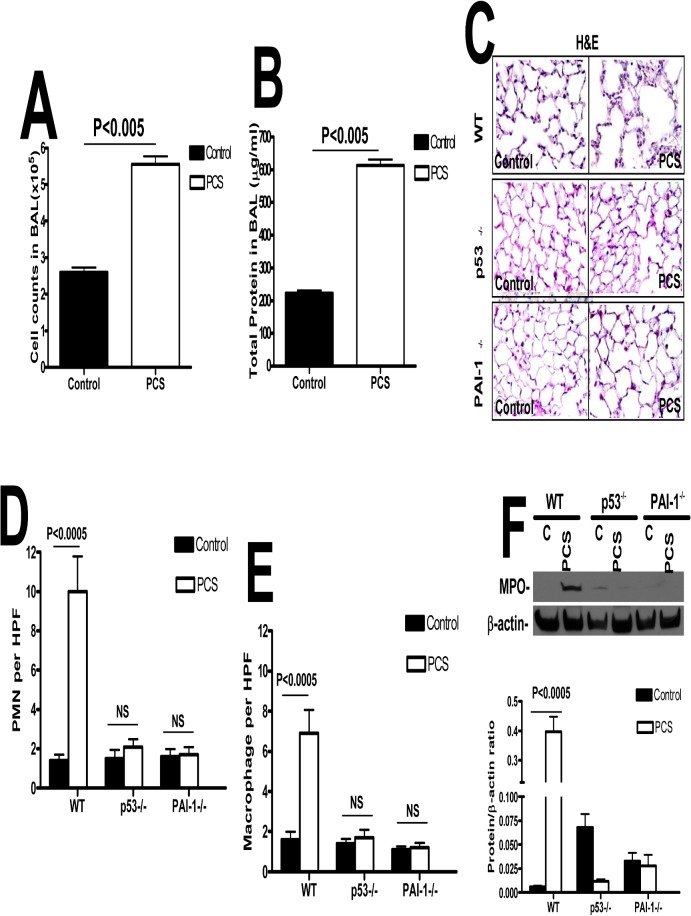
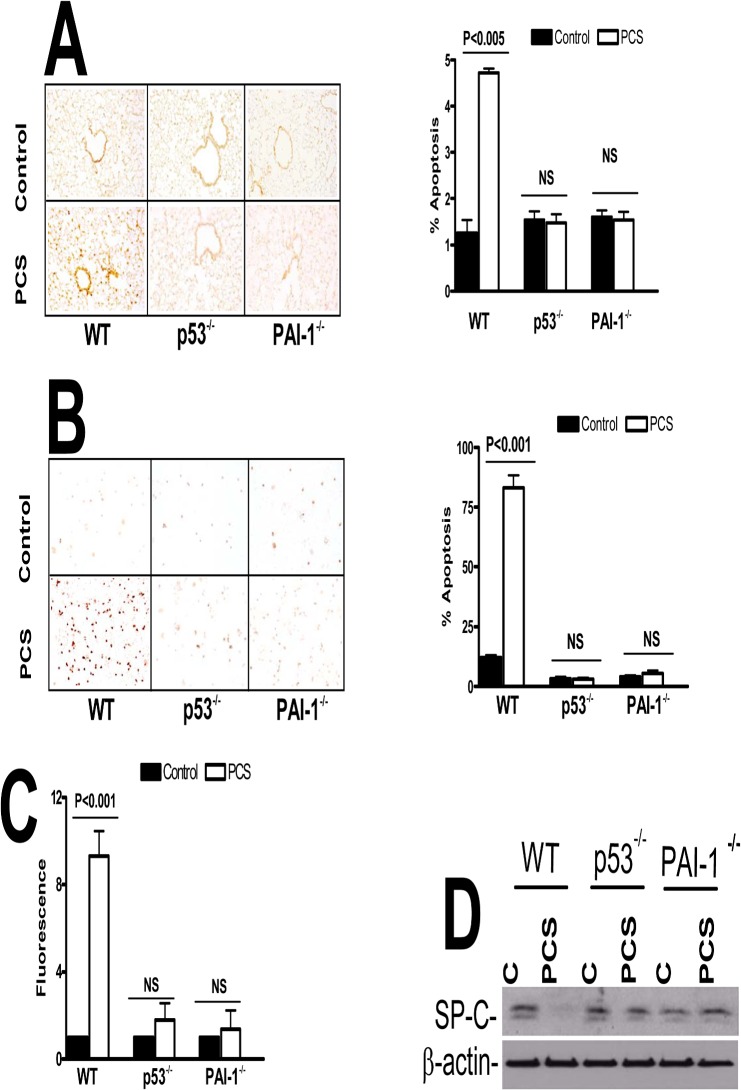
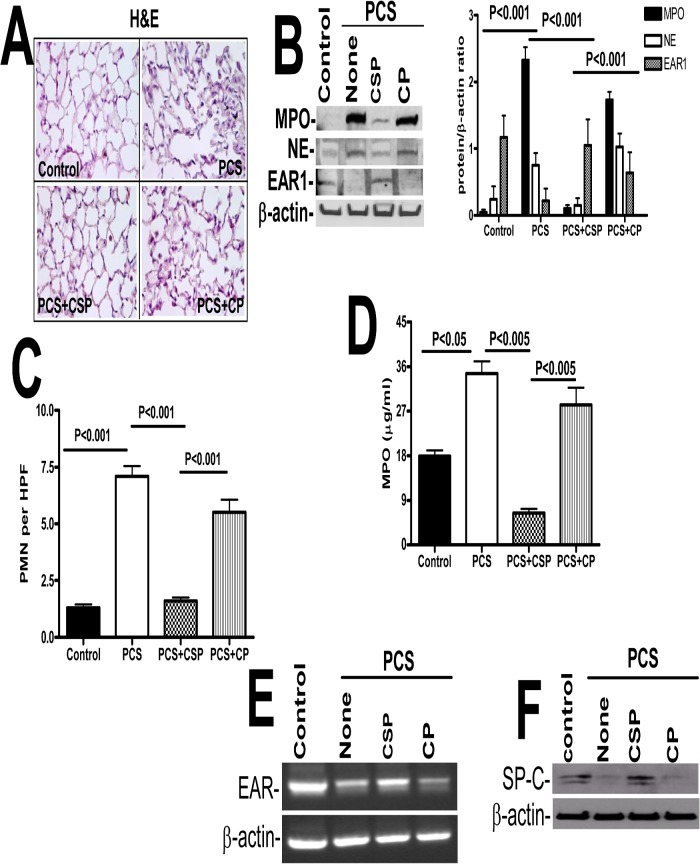
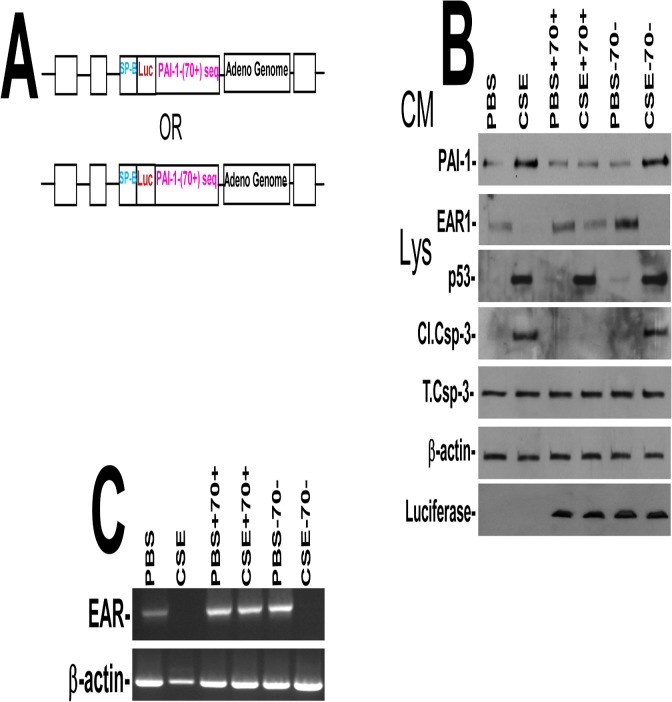
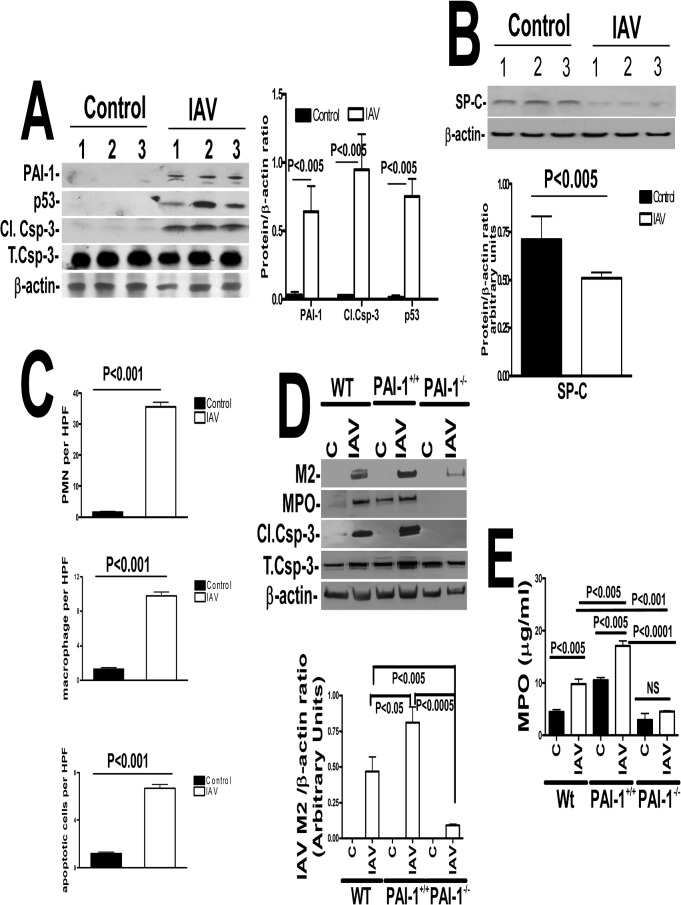
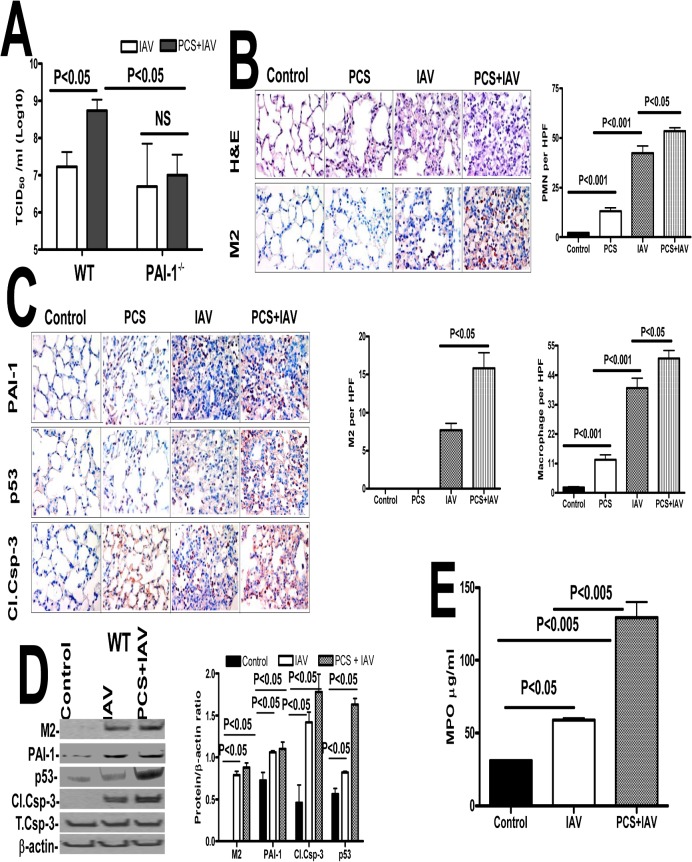
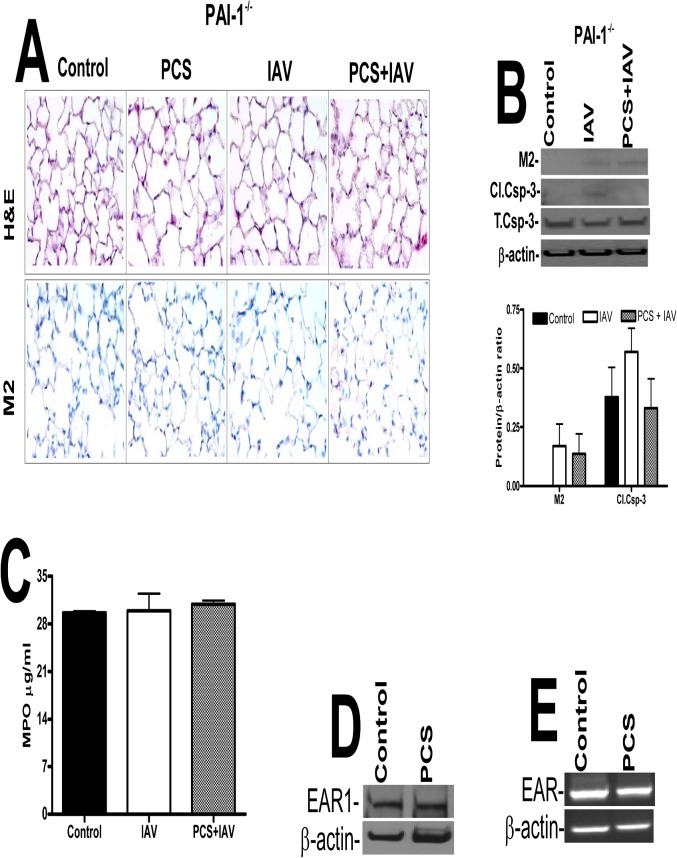
Parenchymal lung inflammation and airway and alveolar epithelial cell apoptosis are associated with cigarette smoke exposure (CSE), which contributes to chronic obstructive pulmonary disease (COPD). Epidemiological studies indicate that people exposed to chronic cigarette smoke with or without COPD are more susceptible to influenza A virus (IAV) infection. We found increased p53, PAI-1 and apoptosis in AECs, with accumulation of macrophages and neutrophils in the lungs of patients with COPD. In Wild-type (WT) mice with passive CSE (PCSE), p53 and PAI-1 expression and apoptosis were increased in AECs as was lung inflammation, while those lacking p53 or PAI-1 resisted AEC apoptosis and lung inflammation. Further, inhibition of p53-mediated induction of PAI-1 by treatment of WT mice with caveolin-1 scaffolding domain peptide (CSP) reduced PCSE-induced lung inflammation and reversed PCSE-induced suppression of eosinophil-associated RNase1 (EAR1). Competitive inhibition of the p53-PAI-1 mRNA interaction by expressing p53-binding 3'UTR sequences of PAI-1 mRNA likewise suppressed CS-induced PAI-1 and AEC apoptosis and restored EAR1 expression. Consistent with PCSE-induced lung injury, IAV infection increased p53, PAI-1 and apoptosis in AECs in association with pulmonary inflammation. Lung inflammation induced by PCSE was worsened by subsequent exposure to IAV. Mice lacking PAI-1 that were exposed to IAV showed minimal viral burden based on M2 antigen and hemagglutination analyses, whereas transgenic mice that overexpress PAI-1 without PCSE showed increased M2 antigen and inflammation after IAV infection. These observations indicate that increased PAI-1 expression promotes AEC apoptosis and exacerbates lung inflammation induced by IAV following PCSE.
Conflict of interest statement
Figures
Similar articles
-
Regulation of airway and alveolar epithelial cell apoptosis by p53-Induced plasminogen activator inhibitor-1 during cigarette smoke exposure injury.Am J Respir Cell Mol Biol. 2012 Oct;47(4):474-83. doi: 10.1165/rcmb.2011-0390OC. Epub 2012 May 16. Am J Respir Cell Mol Biol. 2012. PMID: 22592924 Free PMC article.
-
Caveolin-1-derived peptide attenuates cigarette smoke-induced airway and alveolar epithelial injury.Am J Physiol Lung Cell Mol Physiol. 2023 Nov 1;325(5):L689-L708. doi: 10.1152/ajplung.00178.2022. Epub 2023 Aug 29. Am J Physiol Lung Cell Mol Physiol. 2023. PMID: 37642665 Free PMC article.
-
p53- and PAI-1-mediated induction of C-X-C chemokines and CXCR2: importance in pulmonary inflammation due to cigarette smoke exposure.Am J Physiol Lung Cell Mol Physiol. 2016 Mar 15;310(6):L496-506. doi: 10.1152/ajplung.00290.2015. Epub 2016 Jan 8. Am J Physiol Lung Cell Mol Physiol. 2016. PMID: 26747783 Free PMC article.
-
The induction and consequences of Influenza A virus-induced cell death.Cell Death Dis. 2018 Sep 25;9(10):1002. doi: 10.1038/s41419-018-1035-6. Cell Death Dis. 2018. PMID: 30254192 Free PMC article. Review.
-
In Vivo and In Vitro Studies of Cigarette Smoke Effects on Innate Responses to Influenza Virus: A Matter of Models?Viruses. 2022 Aug 20;14(8):1824. doi: 10.3390/v14081824. Viruses. 2022. PMID: 36016446 Free PMC article. Review.
Cited by
-
Elevated circulating PAI-1 levels are related to lung function decline, systemic inflammation, and small airway obstruction in chronic obstructive pulmonary disease.Int J Chron Obstruct Pulmon Dis. 2016 Sep 26;11:2369-2376. doi: 10.2147/COPD.S107409. eCollection 2016. Int J Chron Obstruct Pulmon Dis. 2016. PMID: 27713627 Free PMC article.
-
Regulation of p53-mediated changes in the uPA-fibrinolytic system and in lung injury by loss of surfactant protein C expression in alveolar epithelial cells.Am J Physiol Lung Cell Mol Physiol. 2017 Jun 1;312(6):L783-L796. doi: 10.1152/ajplung.00291.2016. Epub 2017 Apr 6. Am J Physiol Lung Cell Mol Physiol. 2017. PMID: 28385810 Free PMC article.
-
PAI-1 gain-of-function genotype, factors increasing PAI-1 levels, and airway obstruction: The GALA II Cohort.Clin Exp Allergy. 2017 Sep;47(9):1150-1158. doi: 10.1111/cea.12958. Epub 2017 Jul 3. Clin Exp Allergy. 2017. PMID: 28543872 Free PMC article.
-
Redox control in the pathophysiology of influenza virus infection.BMC Microbiol. 2020 Jul 20;20(1):214. doi: 10.1186/s12866-020-01890-9. BMC Microbiol. 2020. PMID: 32689931 Free PMC article. Review.
-
SERPINE1 -844 and -675 polymorphisms and chronic obstructive pulmonary disease in a Chinese Han population.J Int Med Res. 2016 Dec;44(6):1292-1301. doi: 10.1177/0300060516664270. Epub 2016 Nov 18. J Int Med Res. 2016. PMID: 27856929 Free PMC article.
References
-
- Doll R, Peto R. The causes of cancer: quantitative estimates of avoidable risks of cancer in the United States today. J Natl Cancer Inst. 1981; 66:1191–1308. - PubMed
-
- Jinot J, Bayard S. Respiratory health effects of passive smoking: EPA's weight-of-evidence analysis. J Clin Epidemiol. 1994; 47:339–349. - PubMed
-
- Sopori M. Effects of cigarette smoke on the immune system. Nat. Rev. Immunol. 2002; 2:372–377. - PubMed
-
- Kotani N, Kushikata T, Hashimoto H, Sessler DI, Muraoka M, Matsuki A. Recovery of intraoperative microbicidal and inflammatory functions of alveolar immune cells after a tobacco smoke-free period. Anesthesiology. 2001; 94(6):999–1006. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
