

Predicting human genetic interactions from cancer genome evolution
- PMID: 25933428
- PMCID: PMC4416779
- DOI: 10.1371/journal.pone.0125795
Predicting human genetic interactions from cancer genome evolution
Abstract
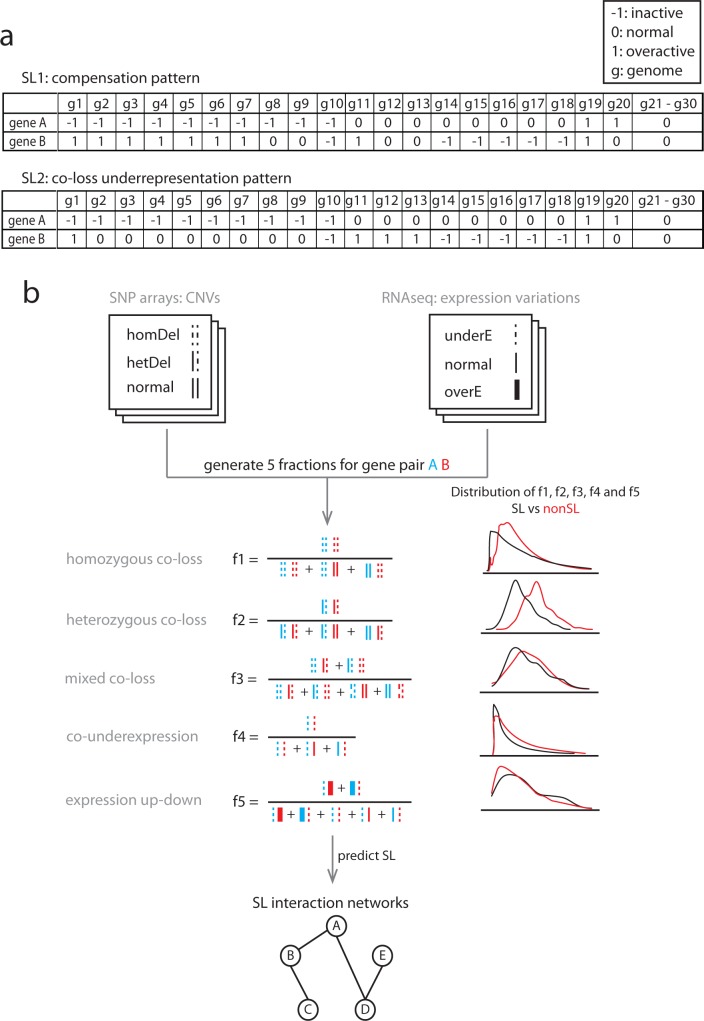
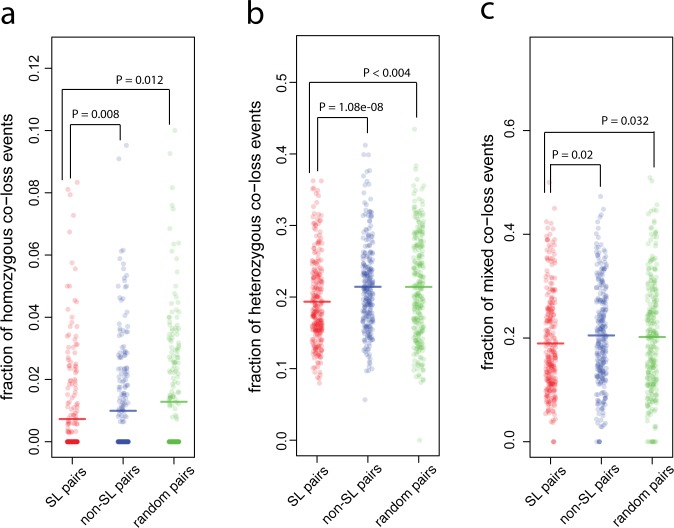
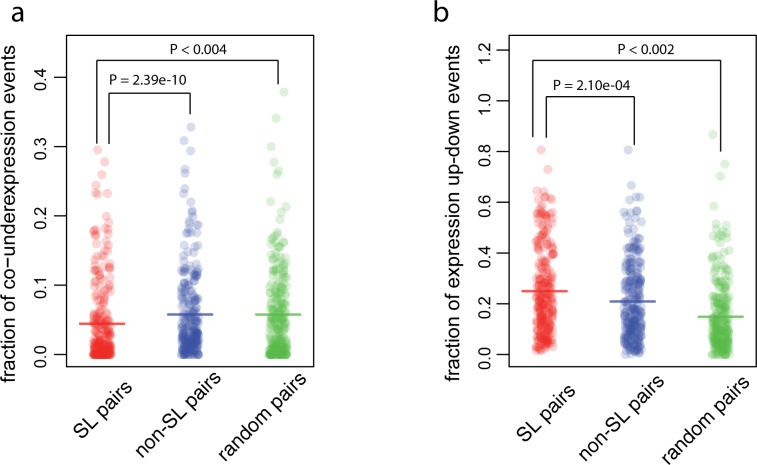
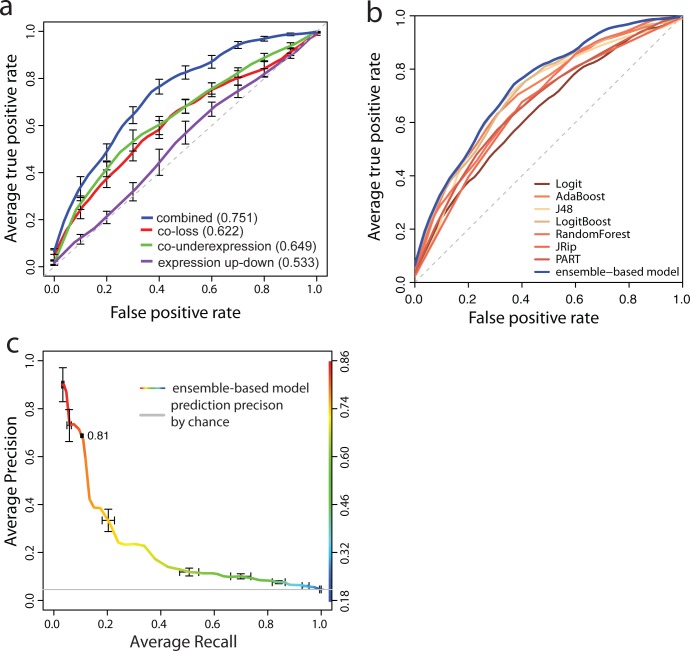
Synthetic Lethal (SL) genetic interactions play a key role in various types of biological research, ranging from understanding genotype-phenotype relationships to identifying drug-targets against cancer. Despite recent advances in empirical measuring SL interactions in human cells, the human genetic interaction map is far from complete. Here, we present a novel approach to predict this map by exploiting patterns in cancer genome evolution. First, we show that empirically determined SL interactions are reflected in various gene presence, absence, and duplication patterns in hundreds of cancer genomes. The most evident pattern that we discovered is that when one member of an SL interaction gene pair is lost, the other gene tends not to be lost, i.e. the absence of co-loss. This observation is in line with expectation, because the loss of an SL interacting pair will be lethal to the cancer cell. SL interactions are also reflected in gene expression profiles, such as an under representation of cases where the genes in an SL pair are both under expressed, and an over representation of cases where one gene of an SL pair is under expressed, while the other one is over expressed. We integrated the various previously unknown cancer genome patterns and the gene expression patterns into a computational model to identify SL pairs. This simple, genome-wide model achieves a high prediction power (AUC = 0.75) for known genetic interactions. It allows us to present for the first time a comprehensive genome-wide list of SL interactions with a high estimated prediction precision, covering up to 591,000 gene pairs. This unique list can potentially be used in various application areas ranging from biotechnology to medical genetics.
Conflict of interest statement
Figures
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
