

Identification of Distinct Conformations of the Angiotensin-II Type 1 Receptor Associated with the Gq/11 Protein Pathway and the β-Arrestin Pathway Using Molecular Dynamics Simulations
- PMID: 25934394
- PMCID: PMC4505491
- DOI: 10.1074/jbc.M114.627356
Identification of Distinct Conformations of the Angiotensin-II Type 1 Receptor Associated with the Gq/11 Protein Pathway and the β-Arrestin Pathway Using Molecular Dynamics Simulations
Abstract
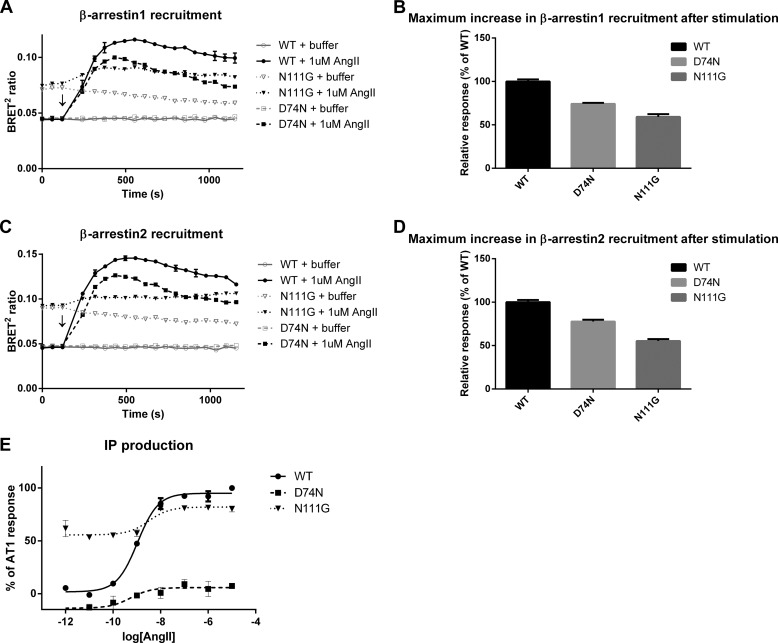
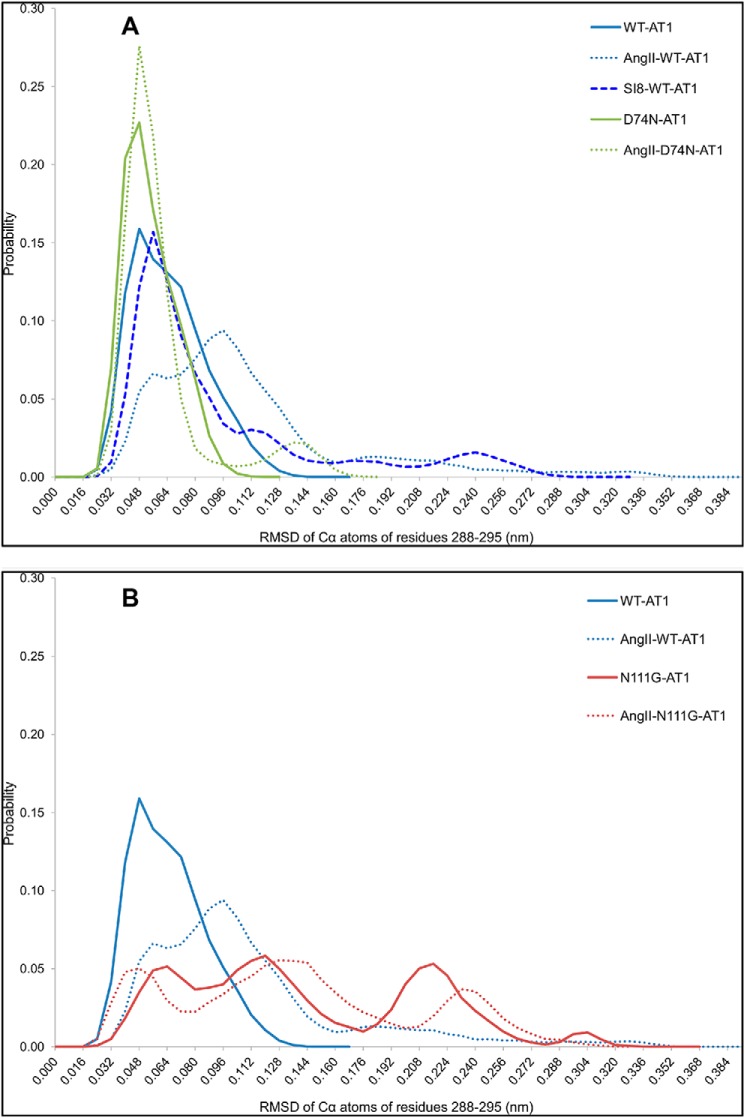
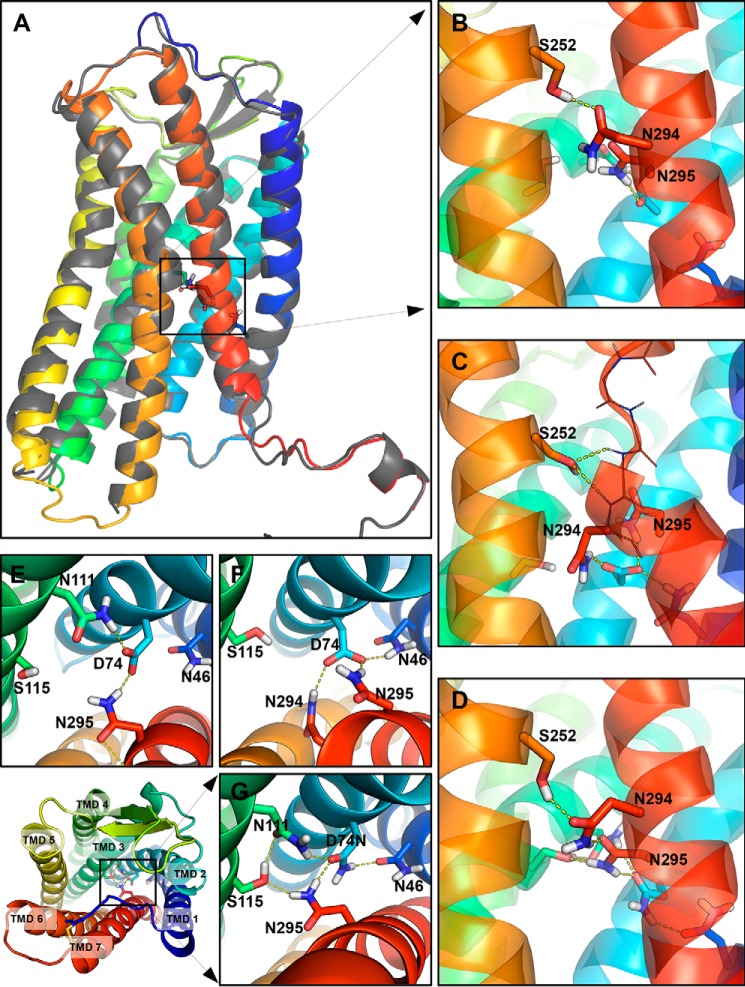
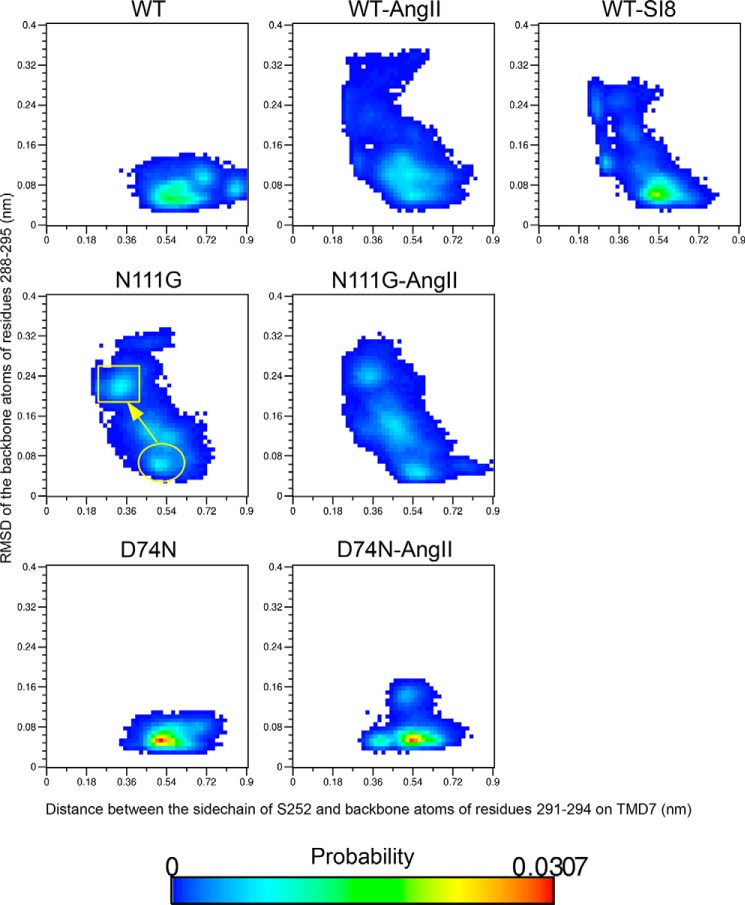
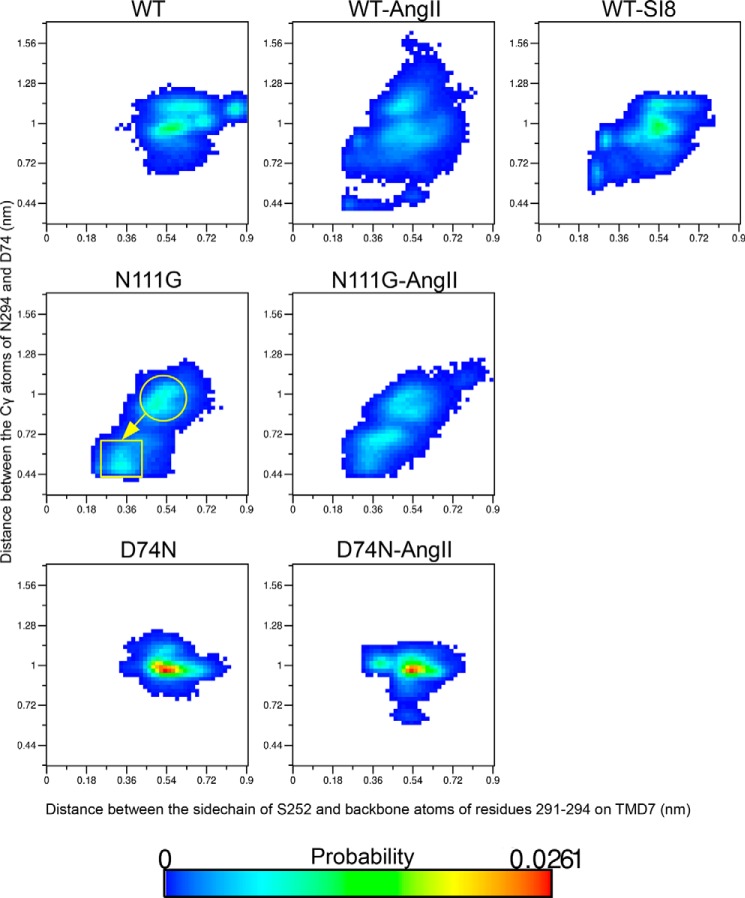
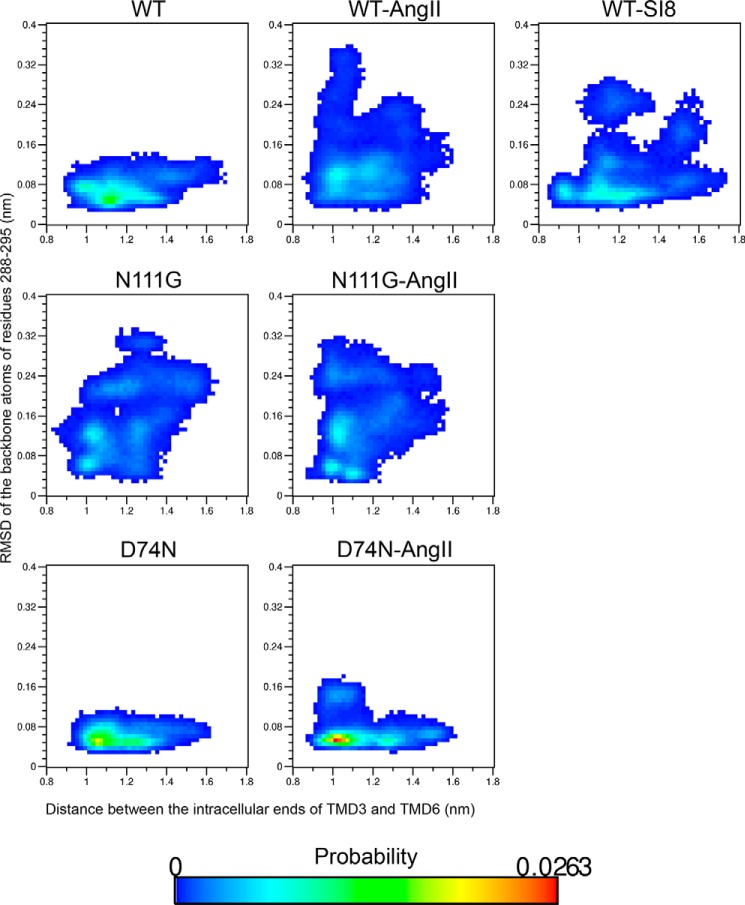
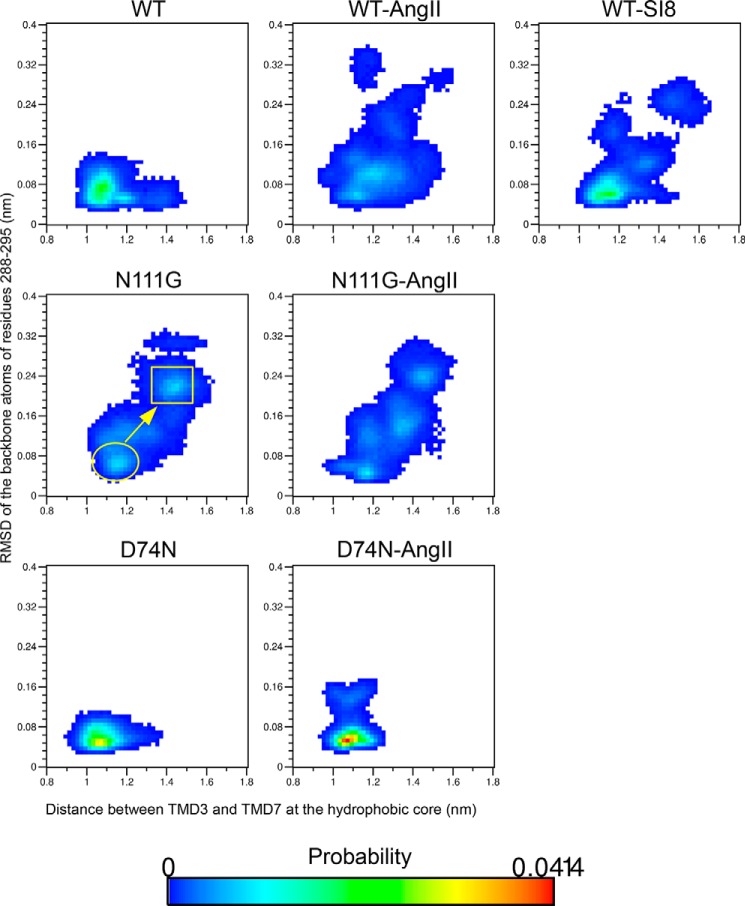
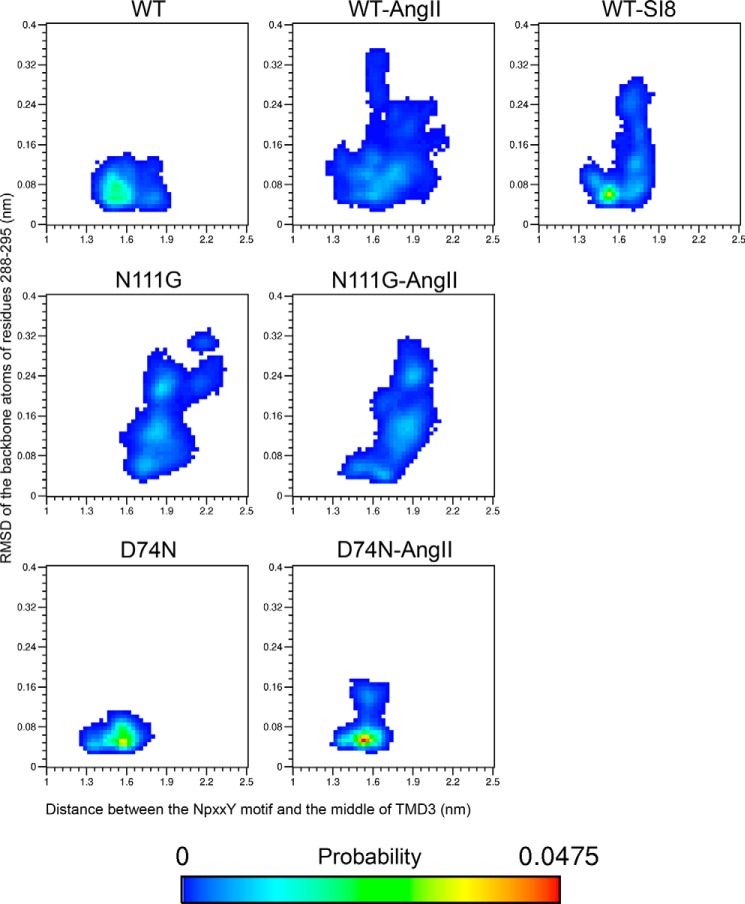
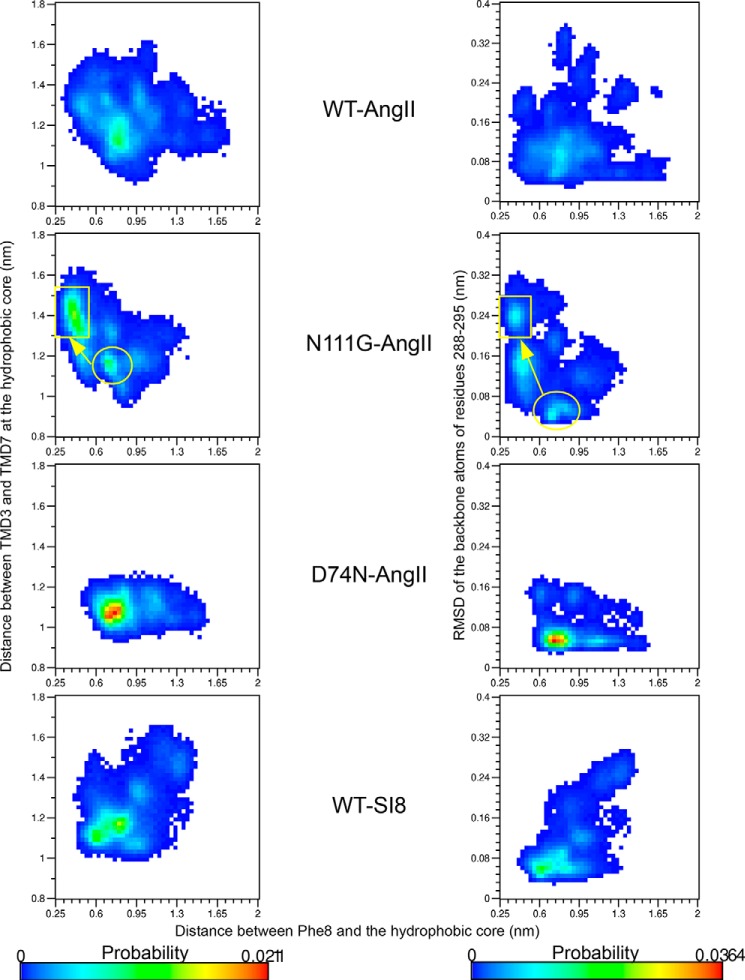
Biased signaling represents the ability of G protein-coupled receptors to engage distinct pathways with various efficacies depending on the ligand used or on mutations in the receptor. The angiotensin-II type 1 (AT1) receptor, a prototypical class A G protein-coupled receptor, can activate various effectors upon stimulation with the endogenous ligand angiotensin-II (AngII), including the Gq/11 protein and β-arrestins. It is believed that the activation of those two pathways can be associated with distinct conformations of the AT1 receptor. To verify this hypothesis, microseconds of molecular dynamics simulations were computed to explore the conformational landscape sampled by the WT-AT1 receptor, the N111G-AT1 receptor (constitutively active and biased for the Gq/11 pathway), and the D74N-AT1 receptor (biased for the β-arrestin1 and -2 pathways) in their apo-forms and in complex with AngII. The molecular dynamics simulations of the AngII-WT-AT1, N111G-AT1, and AngII-N111G-AT1 receptors revealed specific structural rearrangements compared with the initial and ground state of the receptor. Simulations of the D74N-AT1 receptor revealed that the mutation stabilizes the receptor in the initial ground state. The presence of AngII further stabilized the ground state of the D74N-AT1 receptor. The biased agonist [Sar(1),Ile(8)]AngII also showed a preference for the ground state of the WT-AT1 receptor compared with AngII. These results suggest that activation of the Gq/11 pathway is associated with a specific conformational transition stabilized by the agonist, whereas the activation of the β-arrestin pathway is linked to the stabilization of the ground state of the receptor.
Keywords: AT1 receptor; AT1R; G protein-coupled receptor (GPCR); activation mechanism; angiotensin II; biased signaling; functional selectivity; molecular dynamics; protein conformation; receptor structure-function.
© 2015 by The American Society for Biochemistry and Molecular Biology, Inc.
Figures
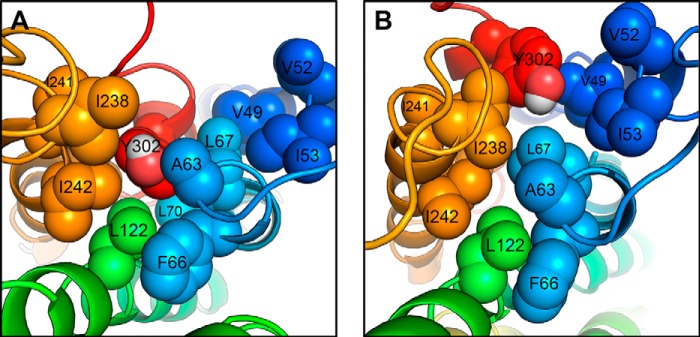
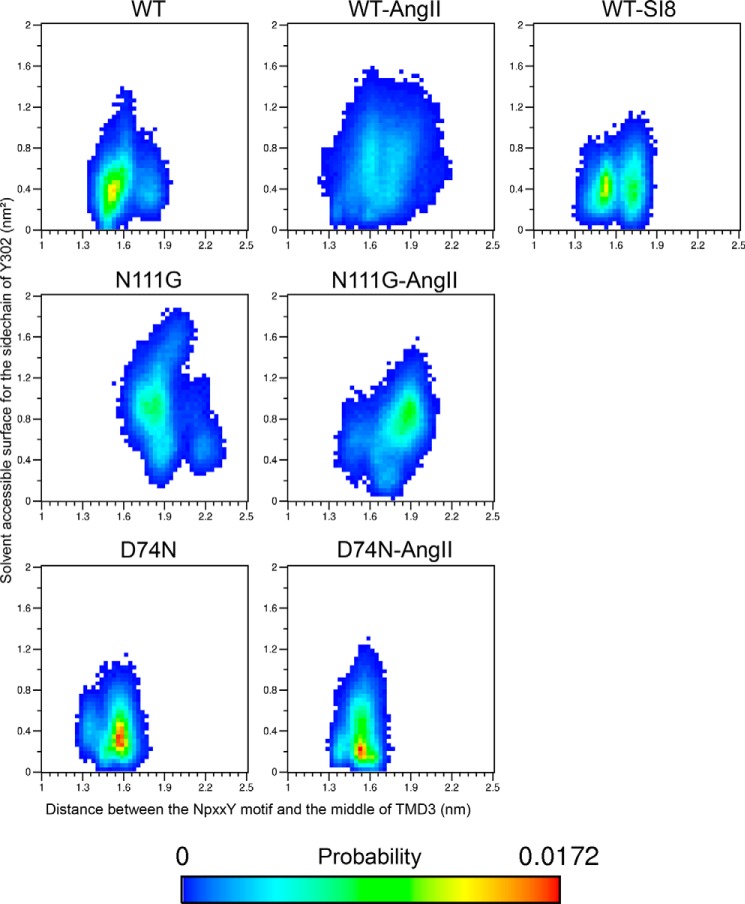
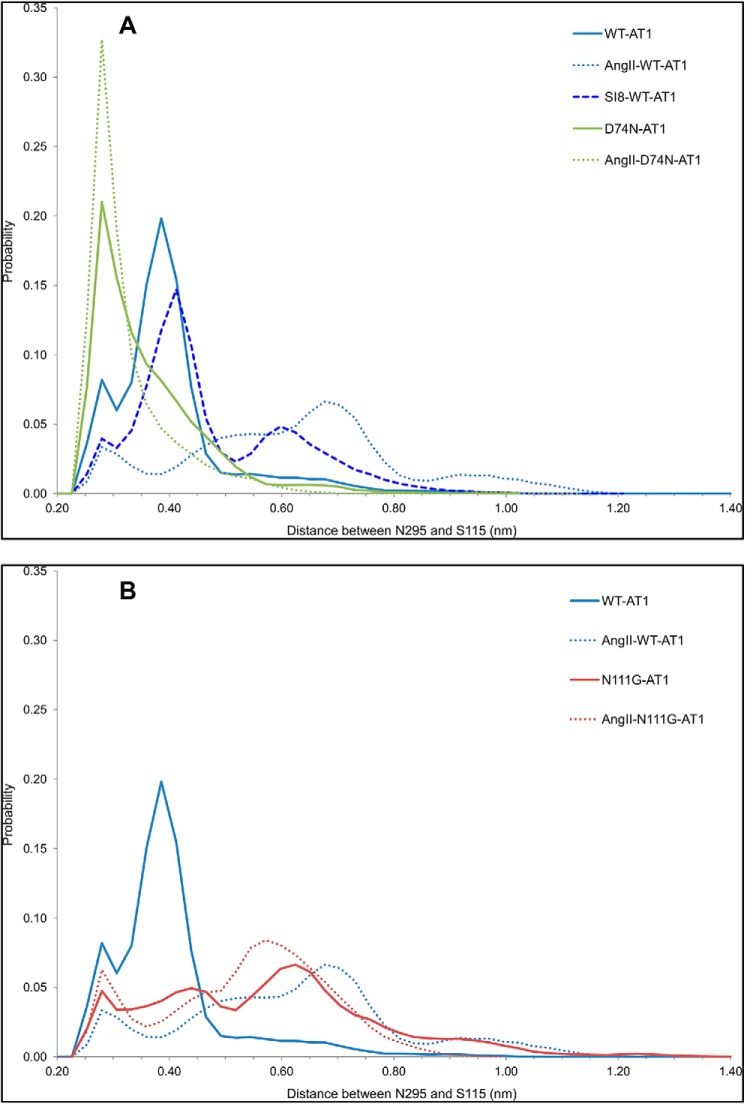
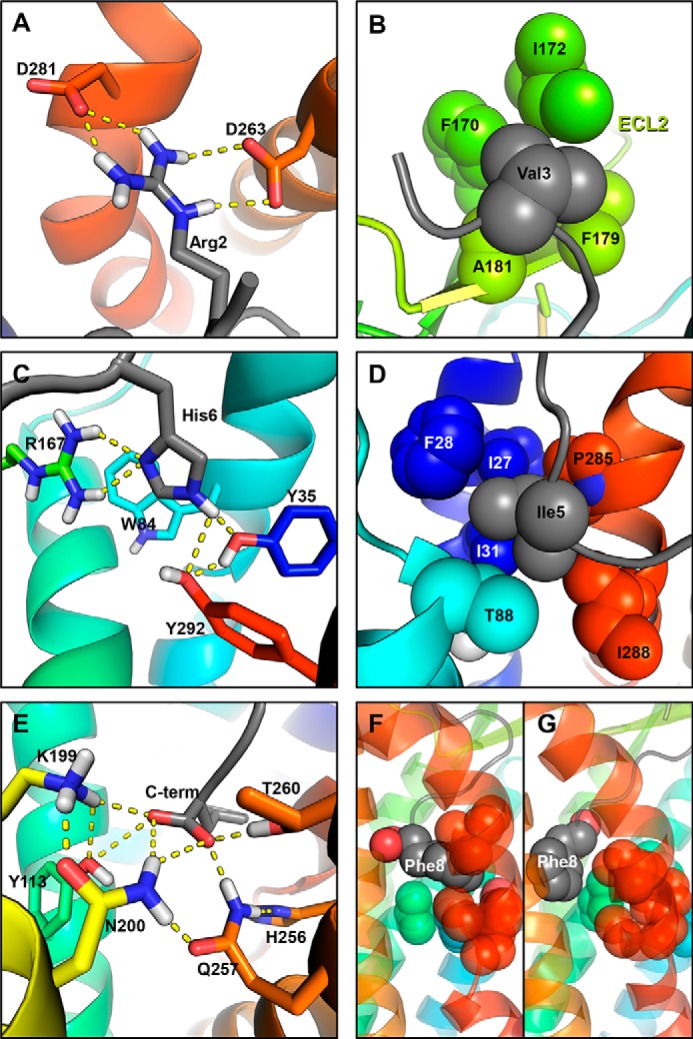
References
-
- de Gasparo M., Catt K. J., Inagami T., Wright J. W., Unger T. (2000) International union of pharmacology. XXIII. The angiotensin II receptors. Pharmacol. Rev. 52, 415–472 - PubMed
-
- Thomas W. G., Qian H. (2003) Arresting angiotensin type 1 receptors. Trends Endocrinol. Metab. 14, 130–136 - PubMed
-
- Aplin M., Bonde M. M., Hansen J. L. (2009) Molecular determinants of angiotensin II type 1 receptor functional selectivity. J. Mol. Cell. Cardiol. 46, 15–24 - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
