

No evidence of locus heterogeneity in familial microcephaly with or without chorioretinopathy, lymphedema, or mental retardation syndrome
- PMID: 25934493
- PMCID: PMC4464120
- DOI: 10.1186/s13023-015-0271-4
No evidence of locus heterogeneity in familial microcephaly with or without chorioretinopathy, lymphedema, or mental retardation syndrome
Abstract
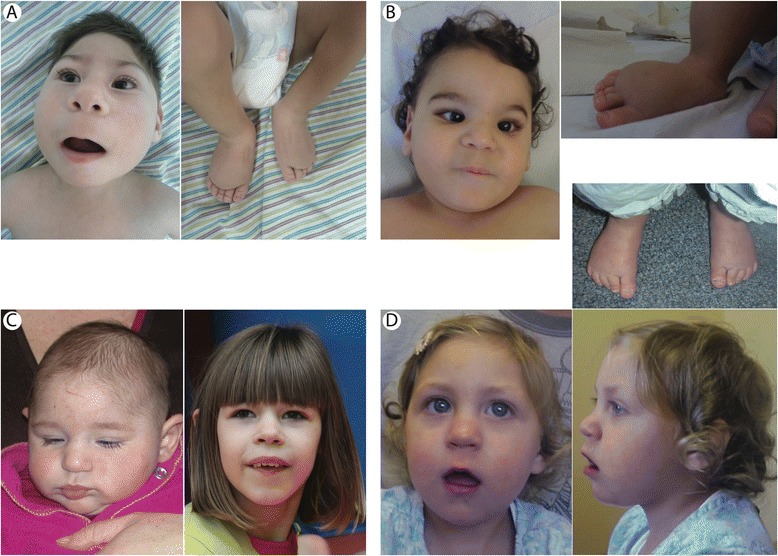
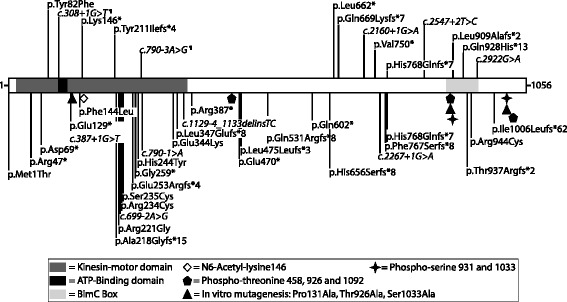
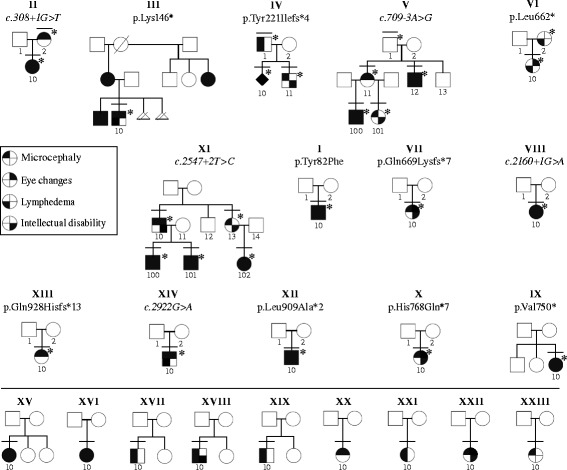
Background: Microcephaly with or without chorioretinopathy, lymphedema, or mental retardation syndrome (MCLMR) is a rare autosomal dominant disorder with variable expressivity. It is characterized by mild-to-severe microcephaly, often associated with intellectual disability, ocular defects and lymphedema. It can be sporadic or inherited. Eighty-seven patients have been described to carry a mutation in KIF11, which encodes a homotetrameric motor kinesin, EG5.
Methods: We tested 23 unreported MCLMR index patients for KIF11. We also reviewed the clinical phenotypes of all our patients as well as of those described in previously published studies.
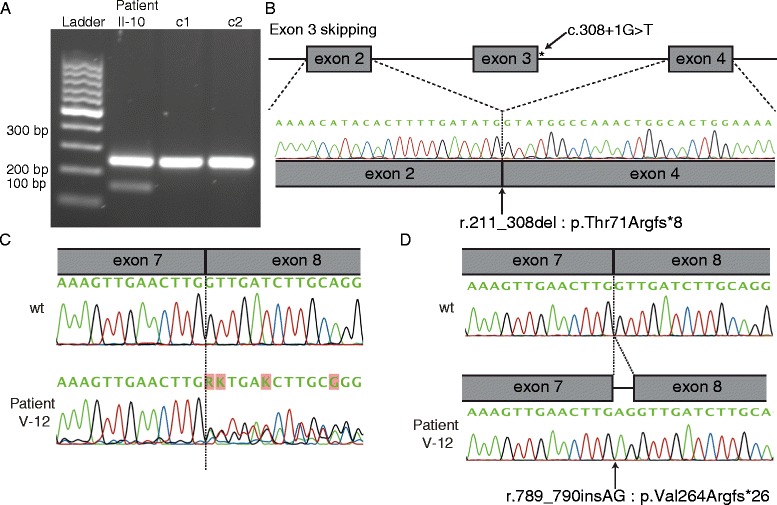
Results: We identified 14 mutations, 12 of which are novel. We detected mutations in 12 affected individuals, from 6 out of 6 familial cases, and in 8 out of 17 sporadic patients. Phenotypic evaluation of patients (our 26 + 61 earlier published = 87) revealed microcephaly in 91%, eye anomalies in 72%, intellectual disability in 67% and lymphedema in 47% of the patients. Unaffected carriers were rare (4 out of 87: 5%). Family history is not a requisite for diagnosis; 31% (16 out of 52) were de novo cases.
Conclusions: All inherited cases, and 50% of sporadic cases of MCLMR are due to germline KIF11 mutations. It is possible that mosaic KIF11 mutations cause the remainder of sporadic cases, which the methods employed here were not designed to detect. On the other hand, some of them might have another mimicking disorder and genetic defect, as microcephaly is highly heterogeneous. In aggregate, KIF11 mutations likely cause the majority, if not all, of MCLMR.
Figures
Similar articles
-
Phenotypic overlap between familial exudative vitreoretinopathy and microcephaly, lymphedema, and chorioretinal dysplasia caused by KIF11 mutations.JAMA Ophthalmol. 2014 Dec;132(12):1393-9. doi: 10.1001/jamaophthalmol.2014.2814. JAMA Ophthalmol. 2014. PMID: 25124931
-
Mutations in KIF11 cause autosomal-dominant microcephaly variably associated with congenital lymphedema and chorioretinopathy.Am J Hum Genet. 2012 Feb 10;90(2):356-62. doi: 10.1016/j.ajhg.2011.12.018. Epub 2012 Jan 26. Am J Hum Genet. 2012. PMID: 22284827 Free PMC article.
-
Congenital microcephaly and chorioretinopathy due to de novo heterozygous KIF11 mutations: five novel mutations and review of the literature.Am J Med Genet A. 2014 Nov;164A(11):2879-86. doi: 10.1002/ajmg.a.36707. Epub 2014 Aug 12. Am J Med Genet A. 2014. PMID: 25115524 Free PMC article. Review.
-
Microcephaly with or without chorioretinopathy, lymphoedema, or mental retardation (MCLMR): review of phenotype associated with KIF11 mutations.Eur J Hum Genet. 2014 Jul;22(7):881-7. doi: 10.1038/ejhg.2013.263. Epub 2013 Nov 27. Eur J Hum Genet. 2014. PMID: 24281367 Free PMC article.
-
Microcephaly, lymphedema, chorioretinal dysplasia (MLCRD) syndrome.J Pediatr Health Care. 2012 Jul-Aug;26(4):306-11. doi: 10.1016/j.pedhc.2011.08.002. Epub 2011 Oct 8. J Pediatr Health Care. 2012. PMID: 22726716 Review. No abstract available.
Cited by
-
An interphase pool of KIF11 localizes at the basal bodies of primary cilia and a reduction in KIF11 expression alters cilia dynamics.Sci Rep. 2020 Aug 18;10(1):13946. doi: 10.1038/s41598-020-70787-4. Sci Rep. 2020. PMID: 32811879 Free PMC article.
-
A Boy With KIF11-Associated Disorder Along With ADHD and ASD: Collaboration Between Paediatrics and Child Psychiatry.Case Rep Psychiatry. 2024 Sep 25;2024:5535830. doi: 10.1155/2024/5535830. eCollection 2024. Case Rep Psychiatry. 2024. PMID: 39359715 Free PMC article.
-
Eg5 and Diseases: From the Well-Known Role in Cancer to the Less-Known Activity in Noncancerous Pathological Conditions.Biochem Res Int. 2024 Jun 20;2024:3649912. doi: 10.1155/2024/3649912. eCollection 2024. Biochem Res Int. 2024. PMID: 38939361 Free PMC article. Review.
-
Identification of a novel mutation in KIF11 with functional analysis in a cohort of 516 familial patients with exudative vitreoretinopathy.Mol Vis. 2021 Sep 1;27:528-541. eCollection 2021. Mol Vis. 2021. PMID: 34526760 Free PMC article.
-
Microcephaly with or without chorioretinopathy, lymphedema, or mental retardation associated with KIF11 pathogenic variant: case report and genotype-phenotype correlation analysis.BMC Ophthalmol. 2025 Jul 30;25(1):437. doi: 10.1186/s12886-025-04261-y. BMC Ophthalmol. 2025. PMID: 40739497 Free PMC article.
References
Publication types
MeSH terms
Substances
Supplementary concepts
LinkOut - more resources
Full Text Sources
Other Literature Sources
