

Second-generation compound for the modulation of utrophin in the therapy of DMD
- PMID: 25935002
- PMCID: PMC4492389
- DOI: 10.1093/hmg/ddv154
Second-generation compound for the modulation of utrophin in the therapy of DMD
Abstract
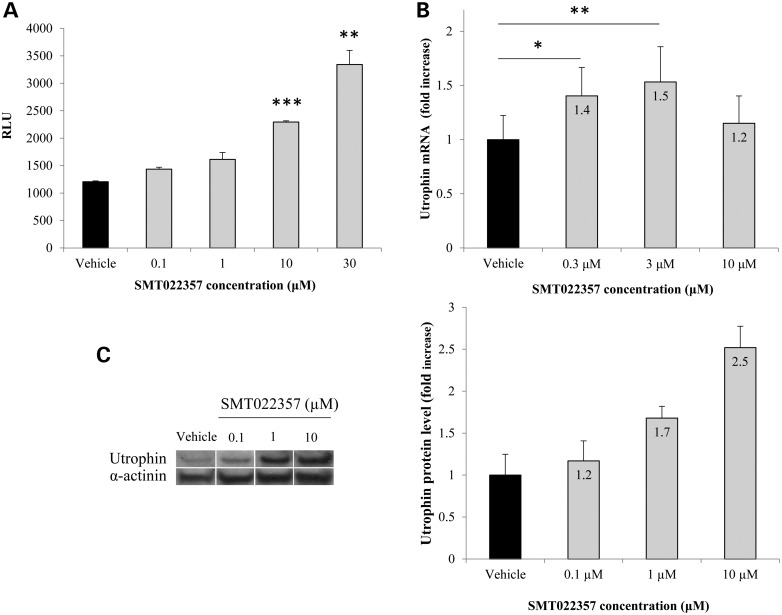
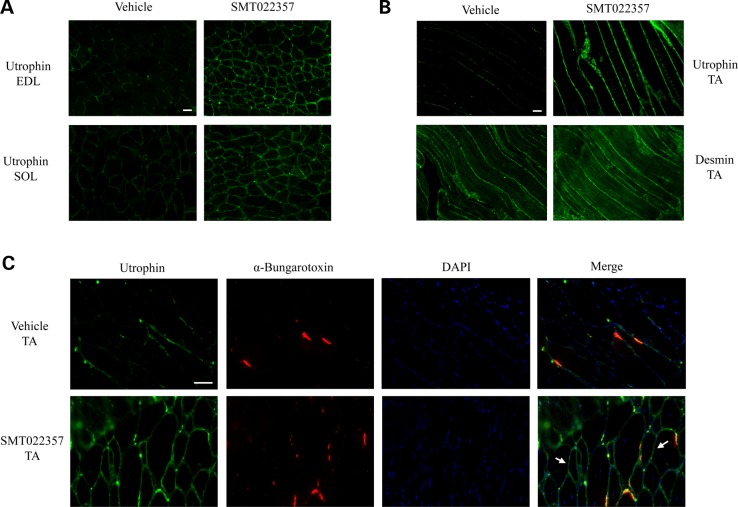
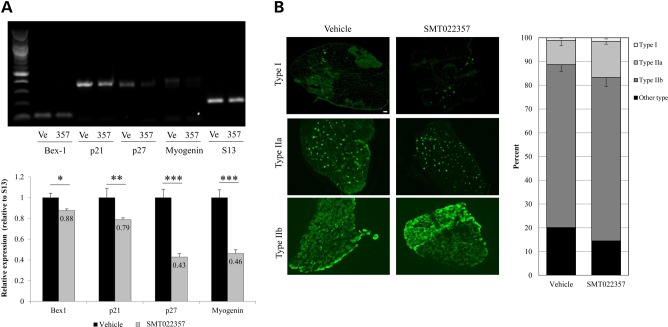
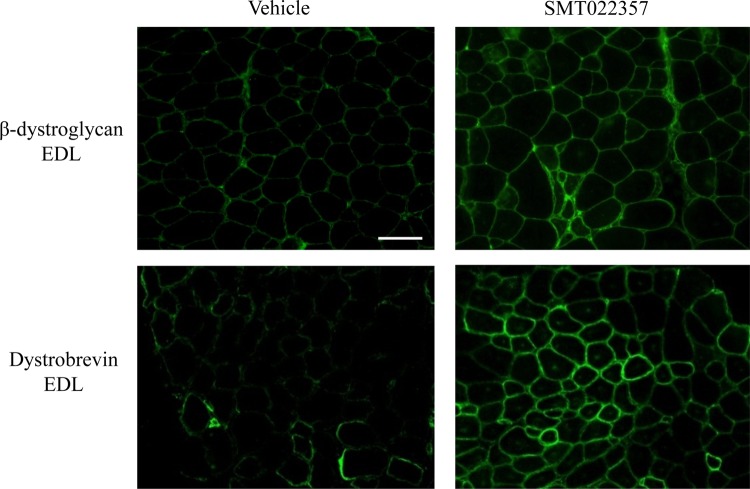
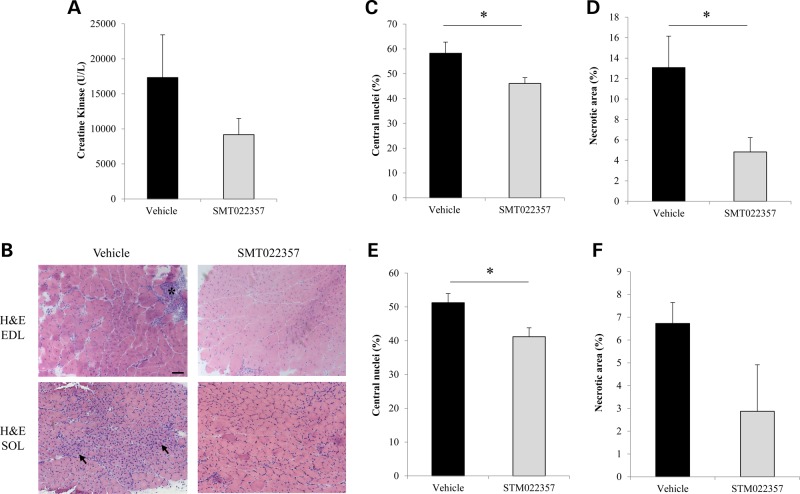
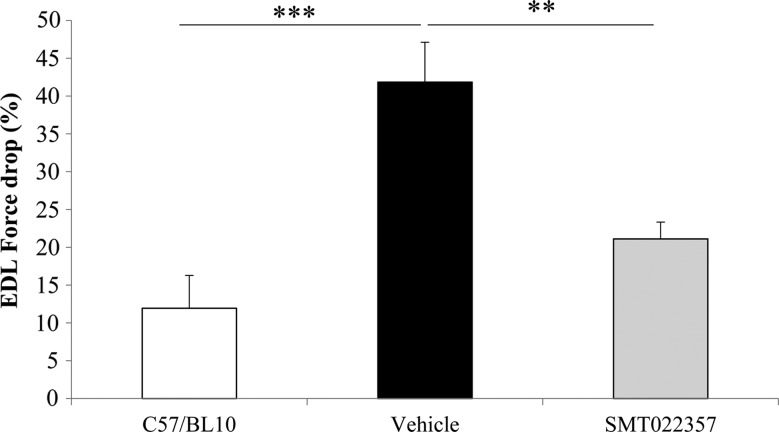
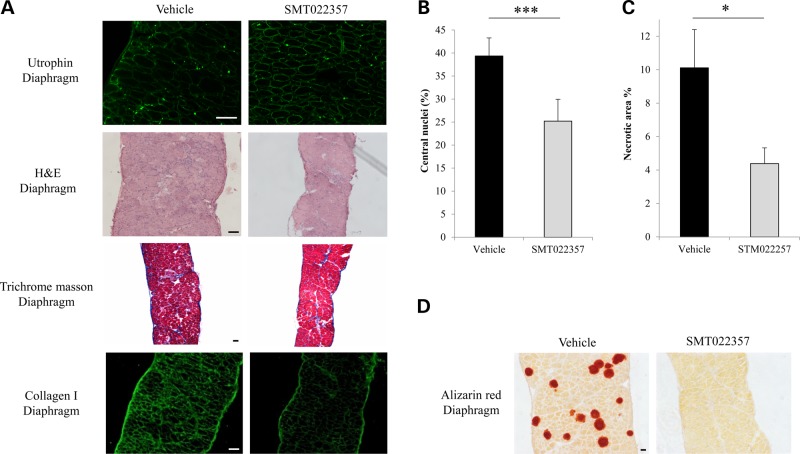
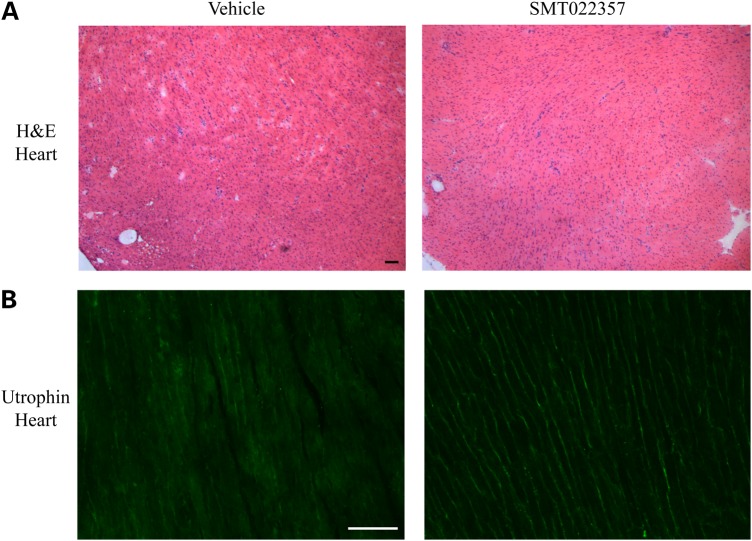
Duchenne muscular dystrophy (DMD) is a lethal, X-linked muscle-wasting disease caused by lack of the cytoskeletal protein dystrophin. There is currently no cure for DMD although various promising approaches are progressing through human clinical trials. By pharmacologically modulating the expression of the dystrophin-related protein utrophin, we have previously demonstrated in dystrophin-deficient mdx studies, daily SMT C1100 treatment significantly reduced muscle degeneration leading to improved muscle function. This manuscript describes the significant disease modifying benefits associated with daily dosing of SMT022357, a second-generation compound in this drug series with improved physicochemical properties and a more robust metabolism profile. These studies in the mdx mouse demonstrate that oral administration of SMT022357 leads to increased utrophin expression in skeletal, respiratory and cardiac muscles. Significantly, utrophin expression is localized along the length of the muscle fibre, not just at the synapse, and is fibre-type independent, suggesting that drug treatment is modulating utrophin transcription in extra-synaptic myonuclei. This results in improved sarcolemmal stability and prevents dystrophic pathology through a significant reduction of regeneration, necrosis and fibrosis. All these improvements combine to protect the mdx muscle from contraction induced damage and enhance physiological function. This detailed evaluation of the SMT C1100 drug series strongly endorses the therapeutic potential of utrophin modulation as a disease modifying therapeutic strategy for all DMD patients irrespective of their dystrophin mutation.
© The Author 2015. Published by Oxford University Press.
Figures
References
-
- Cohn R.D., Campbell K.P. (2000) Molecular basis of muscular dystrophies. Muscle Nerve, 23, 1456–1471. - PubMed
-
- Dickson G., Love D.R., Davies K.E., Wells K.E., Piper T.A., Walsh F.S. (1991) Human dystrophin gene transfer: production and expression of a functional recombinant DNA-based gene. Hum. Genet., 88, 53–58. - PubMed
-
- Emery A.E. (1993) Duchenne muscular dystrophy--Meryon's disease. Neuromusc. Disord., 3, 263–266. - PubMed
-
- Bogdanovich S., Perkins K.J., Krag T.O., Khurana T.S. (2004) Therapeutics for Duchenne muscular dystrophy: current approaches and future directions. J. Mol. Med., 82, 102–115. - PubMed
-
- Bach J.R., O'Brien J., Krotenberg R., Alba A.S. (1987) Management of end stage respiratory failure in Duchenne muscular dystrophy. Muscle Nerve, 10, 177–182. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
