

ATM couples replication stress and metabolic reprogramming during cellular senescence
- PMID: 25937285
- PMCID: PMC4431925
- DOI: 10.1016/j.celrep.2015.04.014
ATM couples replication stress and metabolic reprogramming during cellular senescence
Abstract
Replication stress induced by nucleotide deficiency plays an important role in cancer initiation. Replication stress in primary cells typically activates the cellular senescence tumor-suppression mechanism. Senescence bypass correlates with development of cancer, a disease characterized by metabolic reprogramming. However, the role of metabolic reprogramming in the cellular response to replication stress has been little explored. Here, we report that ataxia telangiectasia mutated (ATM) plays a central role in regulating the cellular response to replication stress by shifting cellular metabolism. ATM inactivation bypasses senescence induced by replication stress triggered by nucleotide deficiency. This was due to restoration of deoxyribonucleotide triphosphate (dNTP) levels through both upregulation of the pentose phosphate pathway via increased glucose-6-phosphate dehydrogenase (G6PD) activity and enhanced glucose and glutamine consumption. These phenotypes were mediated by a coordinated suppression of p53 and upregulation of c-MYC downstream of ATM inactivation. Our data indicate that ATM status couples replication stress and metabolic reprogramming during senescence.
Copyright © 2015 The Authors. Published by Elsevier Inc. All rights reserved.
Figures
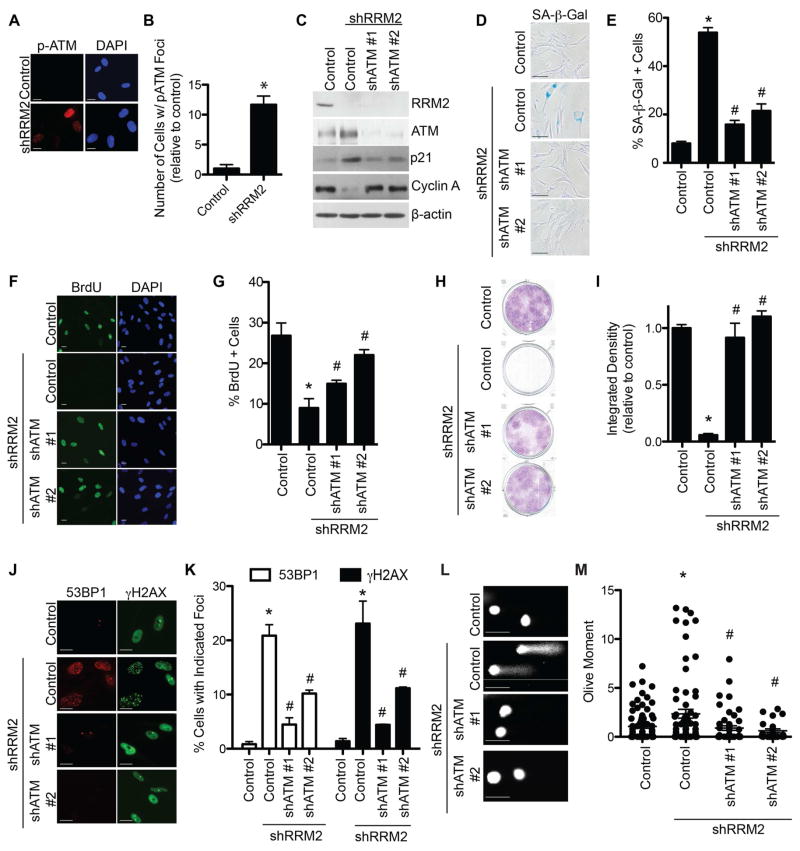
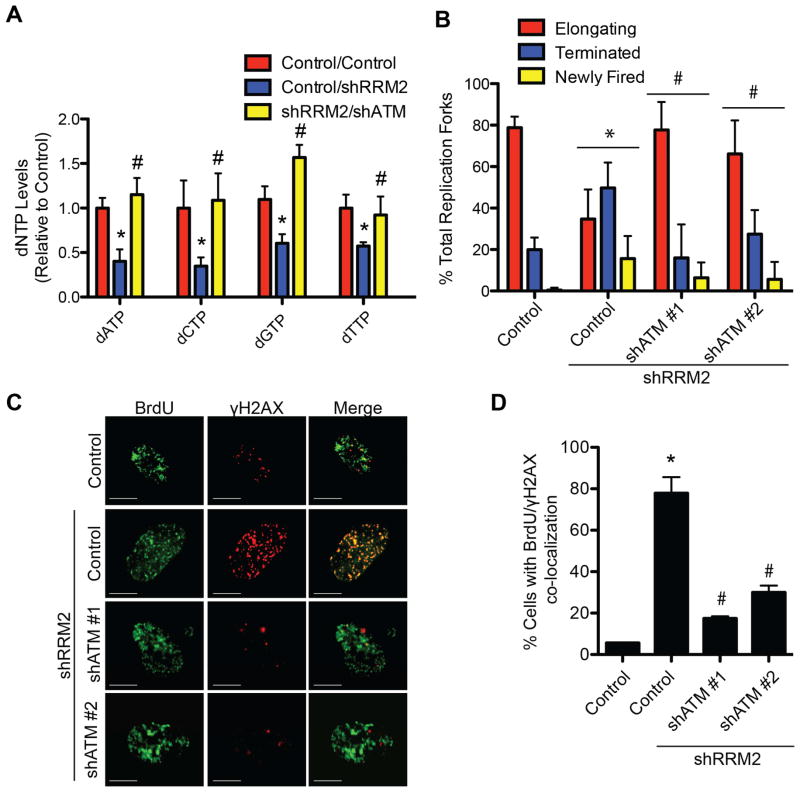
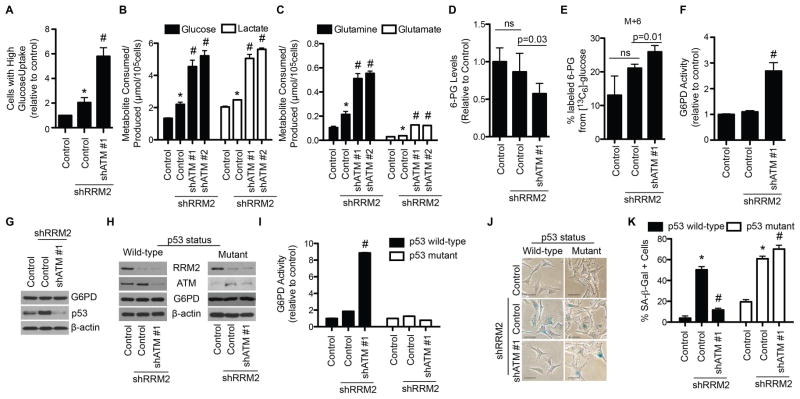
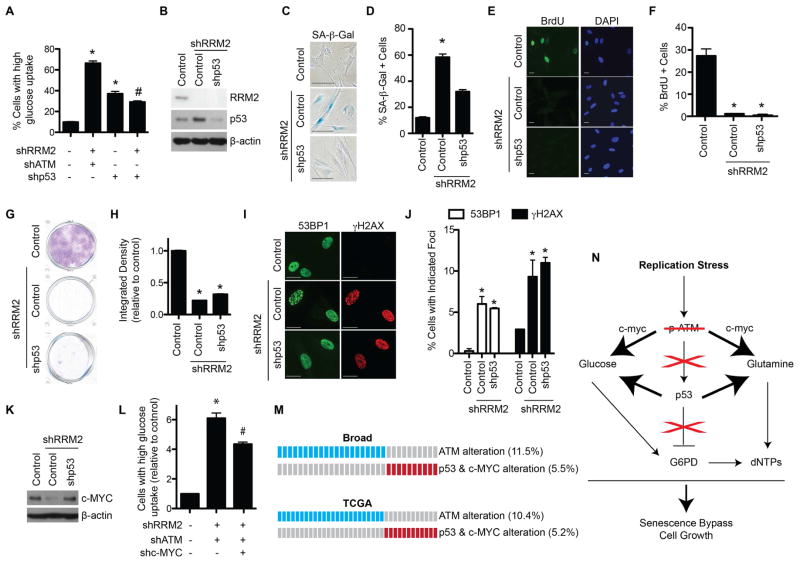
References
-
- Bartkova J, Rezaei N, Liontos M, Karakaidos P, Kletsas D, Issaeva N, Vassiliou LV, Kolettas E, Niforou K, Zoumpourlis VC, et al. Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature. 2006;444:633–637. - PubMed
-
- Blakley RL, Vitols E. The control of nucleotide biosynthesis. Annu Rev Biochem. 1968;37:201–224. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- K99CA194309/CA/NCI NIH HHS/United States
- T32CA9171-35/CA/NCI NIH HHS/United States
- T32 GM008076/GM/NIGMS NIH HHS/United States
- P50CA174523/CA/NCI NIH HHS/United States
- P30 CA010815/CA/NCI NIH HHS/United States
- R00 CA194309/CA/NCI NIH HHS/United States
- K99 CA194309/CA/NCI NIH HHS/United States
- R01 CA174761/CA/NCI NIH HHS/United States
- P30 ES013508/ES/NIEHS NIH HHS/United States
- P50 CA174523/CA/NCI NIH HHS/United States
- R01CA160331/CA/NCI NIH HHS/United States
- T32ES019851/ES/NIEHS NIH HHS/United States
- CA010815/CA/NCI NIH HHS/United States
- P30 CA016520/CA/NCI NIH HHS/United States
- R01 CA160331/CA/NCI NIH HHS/United States
- T32 CA009171/CA/NCI NIH HHS/United States
- T32 ES019851/ES/NIEHS NIH HHS/United States
- R21 CA194973/CA/NCI NIH HHS/United States
- P30ES013508/ES/NIEHS NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
