

Genetic Basis of Common Human Disease: Insight into the Role of Missense SNPs from Genome-Wide Association Studies
- PMID: 25937569
- PMCID: PMC4893807
- DOI: 10.1016/j.jmb.2015.04.014
Genetic Basis of Common Human Disease: Insight into the Role of Missense SNPs from Genome-Wide Association Studies
Abstract
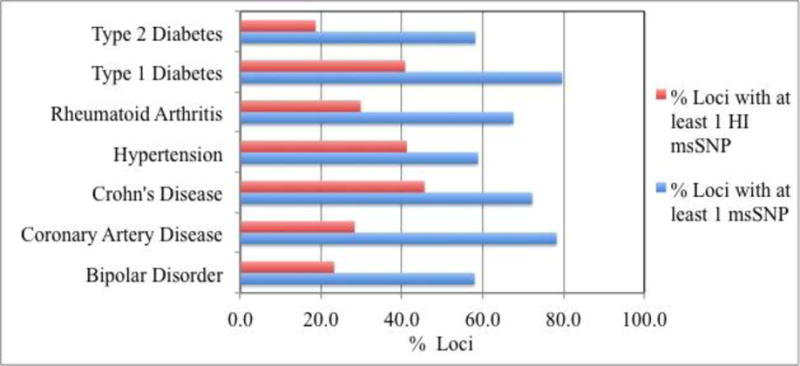
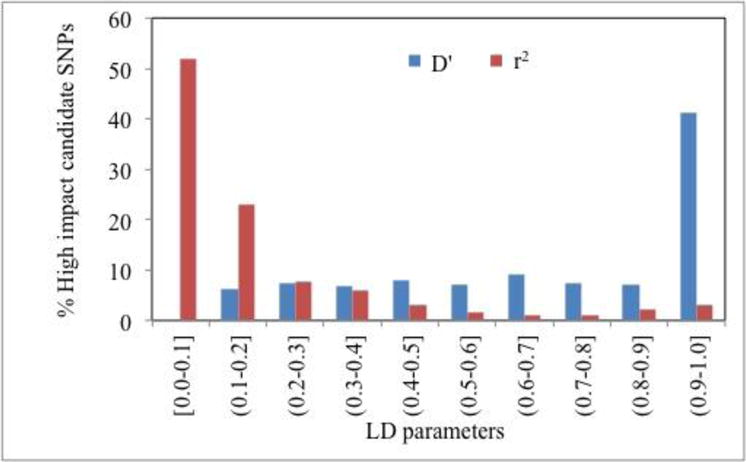
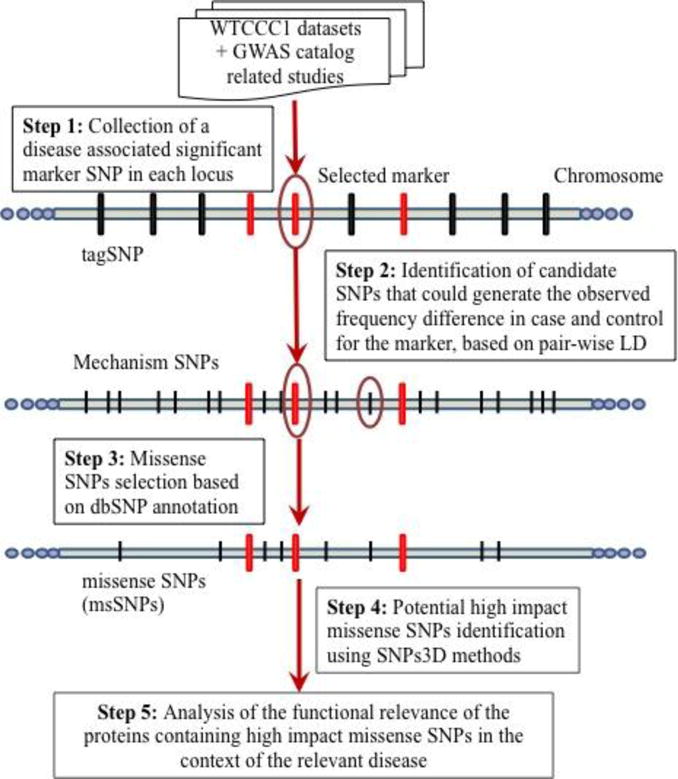
Recent genome-wide association studies (GWAS) have led to the reliable identification of single nucleotide polymorphisms (SNPs) at a number of loci associated with increased risk of specific common human diseases. Each such locus implicates multiple possible candidate SNPs for involvement in disease mechanism. A variety of mechanisms may link the presence of an SNP to altered in vivo gene product function and hence contribute to disease risk. Here, we report an analysis of the role of one of these mechanisms, missense SNPs (msSNPs) in proteins in seven complex trait diseases. Linkage disequilibrium information was used to identify possible candidate msSNPs associated with increased disease risk at each of 356 loci for the seven diseases. Two computational methods were used to estimate which of these SNPs has a significant impact on in vivo protein function. 69% of the loci have at least one candidate msSNP and 33% have at least one predicted high-impact msSNP. In some cases, these SNPs are in well-established disease-related proteins, such as MST1 (macrophage stimulating 1) for Crohn's disease. In others, they are in proteins identified by GWAS as likely candidates for disease relevance, but previously without known mechanism, such as ADAMTS13 (ADAM metallopeptidase with thrombospondin type 1 motif, 13) for coronary artery disease. In still other cases, the missense SNPs are in proteins not previously suggested as disease candidates, such as TUBB1 (tubulin, beta 1, class VI) for hypertension. Together, these data support a substantial role for this class of SNPs in susceptibility to common human disease.
Keywords: GWAS; complex trait disease; nonsynonymous; protein structure.
Copyright © 2015. Published by Elsevier Ltd.
Figures


Similar articles
-
Increasing power of genome-wide association studies by collecting additional single-nucleotide polymorphisms.Genetics. 2011 Jun;188(2):449-60. doi: 10.1534/genetics.111.128595. Epub 2011 Apr 5. Genetics. 2011. PMID: 21467568 Free PMC article.
-
Gene-centric association mapping of chromosome 3p implicates MST1 in IBD pathogenesis.Mucosal Immunol. 2008 Mar;1(2):131-8. doi: 10.1038/mi.2007.15. Epub 2008 Jan 16. Mucosal Immunol. 2008. PMID: 19079170 Free PMC article.
-
Quantifying missing heritability at known GWAS loci.PLoS Genet. 2013;9(12):e1003993. doi: 10.1371/journal.pgen.1003993. Epub 2013 Dec 26. PLoS Genet. 2013. PMID: 24385918 Free PMC article.
-
Shared genetic etiology underlying Alzheimer's disease and type 2 diabetes.Mol Aspects Med. 2015 Jun-Oct;43-44:66-76. doi: 10.1016/j.mam.2015.06.006. Epub 2015 Jun 23. Mol Aspects Med. 2015. PMID: 26116273 Free PMC article. Review.
-
Reducing GWAS Complexity.Cell Cycle. 2016;15(1):22-4. doi: 10.1080/15384101.2015.1120928. Cell Cycle. 2016. PMID: 26771711 Free PMC article. Review.
Cited by
-
Consensus Genome-Wide Expression Quantitative Trait Loci and Their Relationship with Human Complex Trait Disease.OMICS. 2016 Jul;20(7):400-14. doi: 10.1089/omi.2016.0063. OMICS. 2016. PMID: 27428252 Free PMC article.
-
Biophysical and Mechanistic Models for Disease-Causing Protein Variants.Trends Biochem Sci. 2019 Jul;44(7):575-588. doi: 10.1016/j.tibs.2019.01.003. Epub 2019 Jan 31. Trends Biochem Sci. 2019. PMID: 30712981 Free PMC article. Review.
-
A comprehensive in silico investigation into the pathogenic SNPs in the RTEL1 gene and their biological consequences.PLoS One. 2024 Sep 6;19(9):e0309713. doi: 10.1371/journal.pone.0309713. eCollection 2024. PLoS One. 2024. PMID: 39240887 Free PMC article.
-
The gasdermins: a pore-forming protein family expressed in the epidermis.Front Immunol. 2023 Sep 12;14:1254150. doi: 10.3389/fimmu.2023.1254150. eCollection 2023. Front Immunol. 2023. PMID: 37771587 Free PMC article. Review.
-
ACMG/AMP interpretation of BRCA1 missense variants: Structure-informed scores add evidence strength granularity to the PP3/BP4 computational evidence.Am J Hum Genet. 2025 May 1;112(5):993-1002. doi: 10.1016/j.ajhg.2024.12.011. Epub 2025 Apr 14. Am J Hum Genet. 2025. PMID: 40233743 Free PMC article.
References
-
-
dbSNP n.d.
http://www.ncbi.nlm.nih.gov/projects/SNP/ .
-
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
