

Knowledge-guided docking: accurate prospective prediction of bound configurations of novel ligands using Surflex-Dock
- PMID: 25940276
- PMCID: PMC4464052
- DOI: 10.1007/s10822-015-9846-3
Knowledge-guided docking: accurate prospective prediction of bound configurations of novel ligands using Surflex-Dock
Abstract
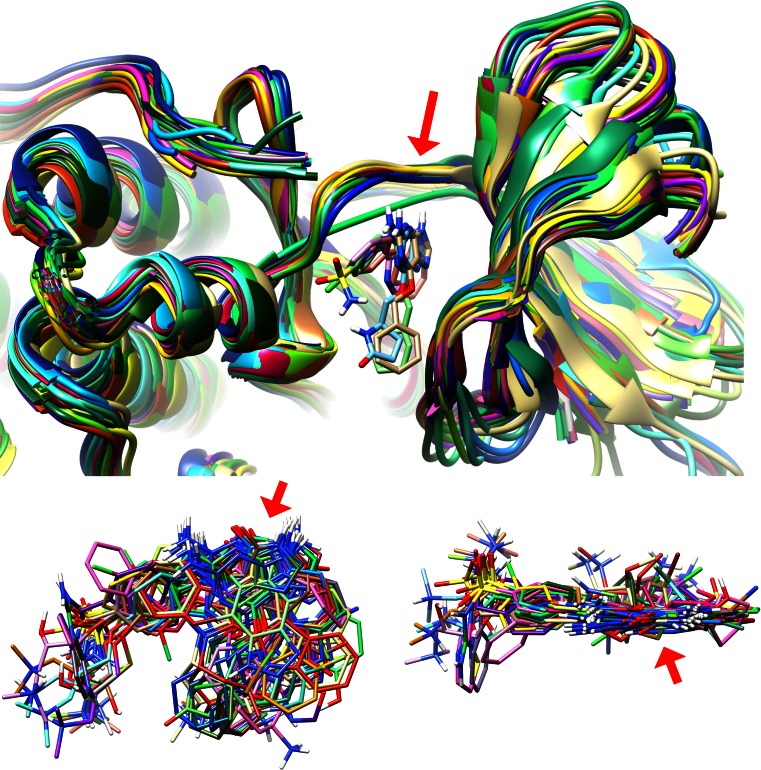
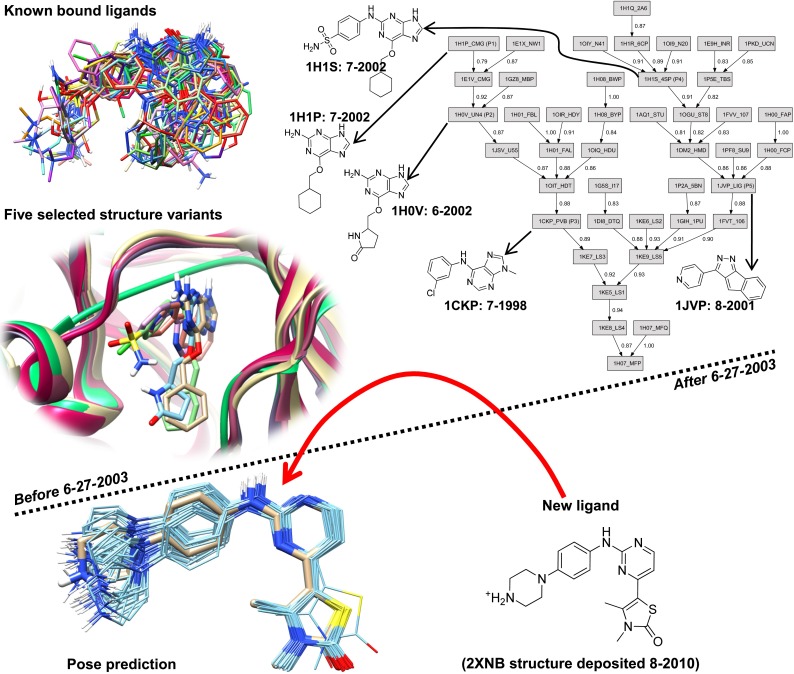
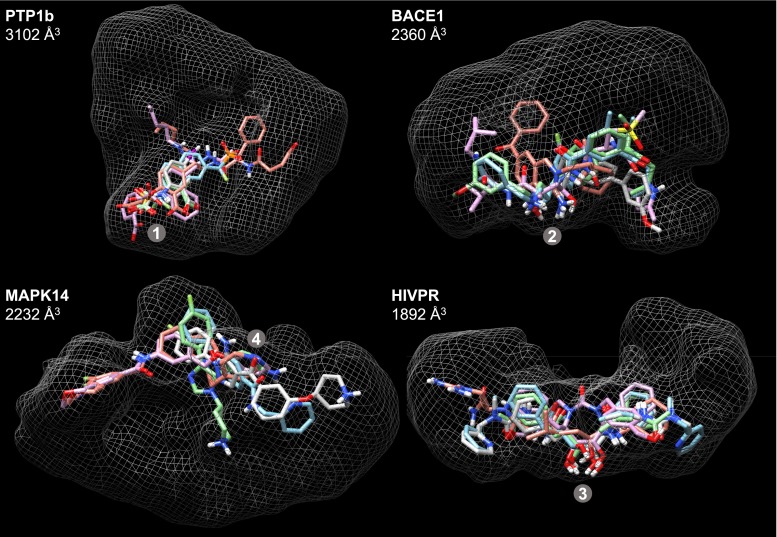
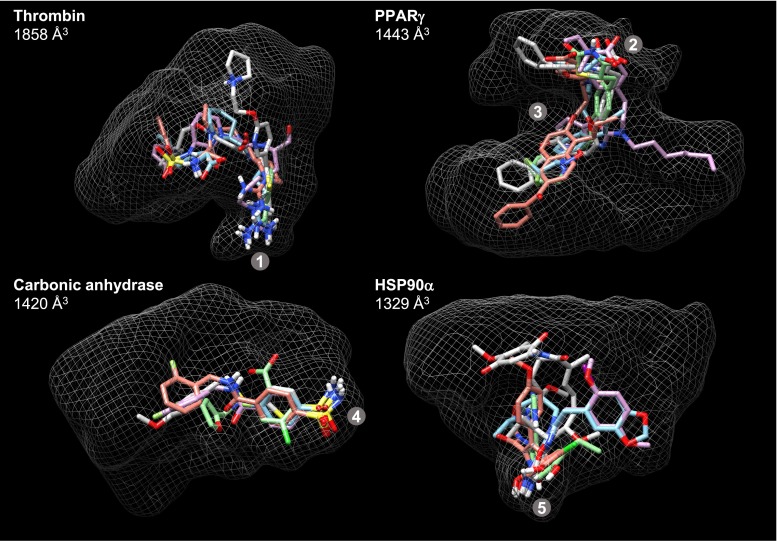
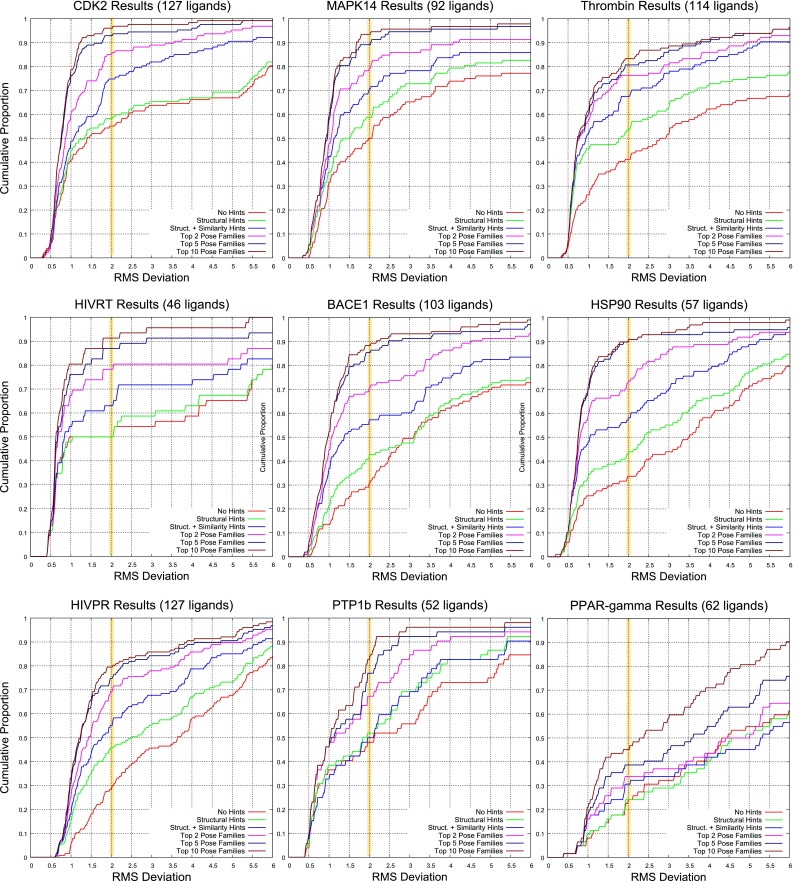
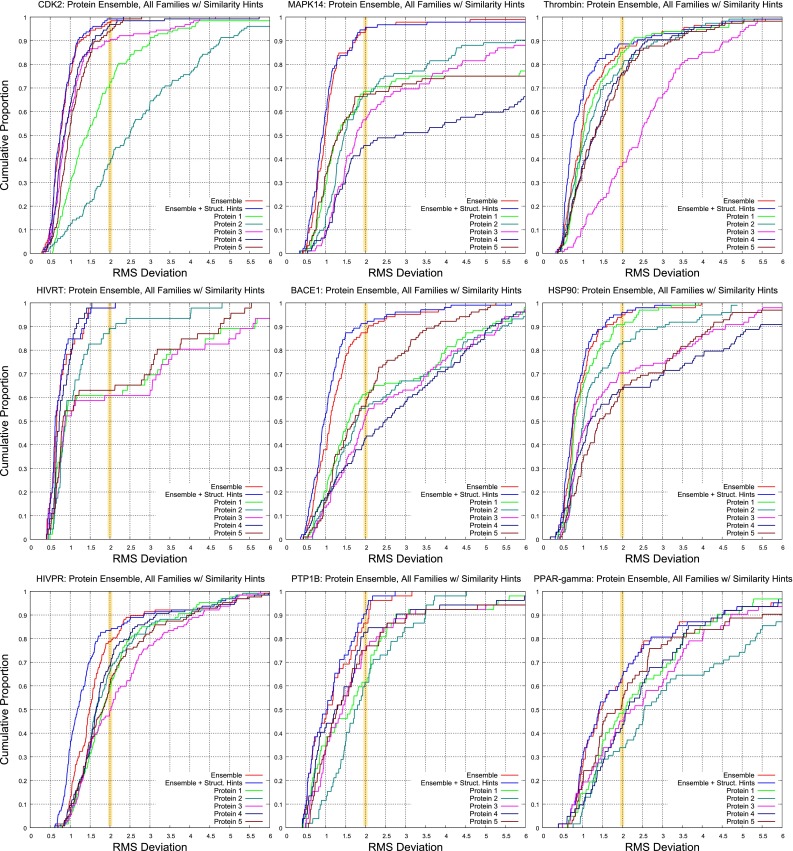
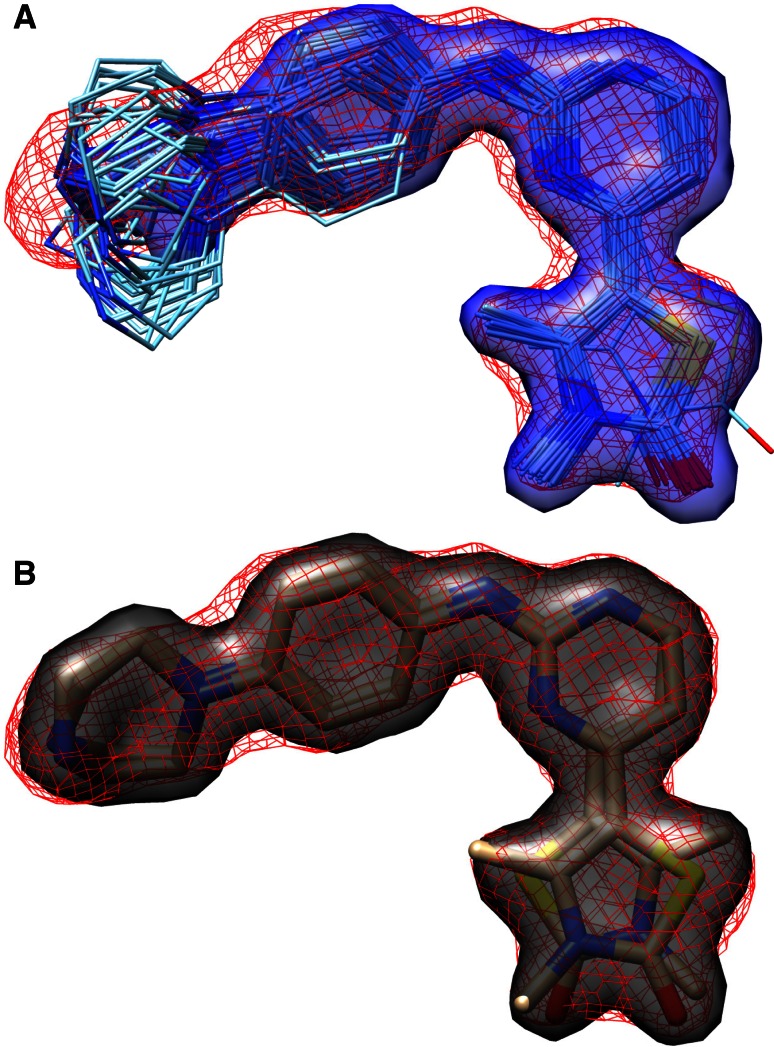
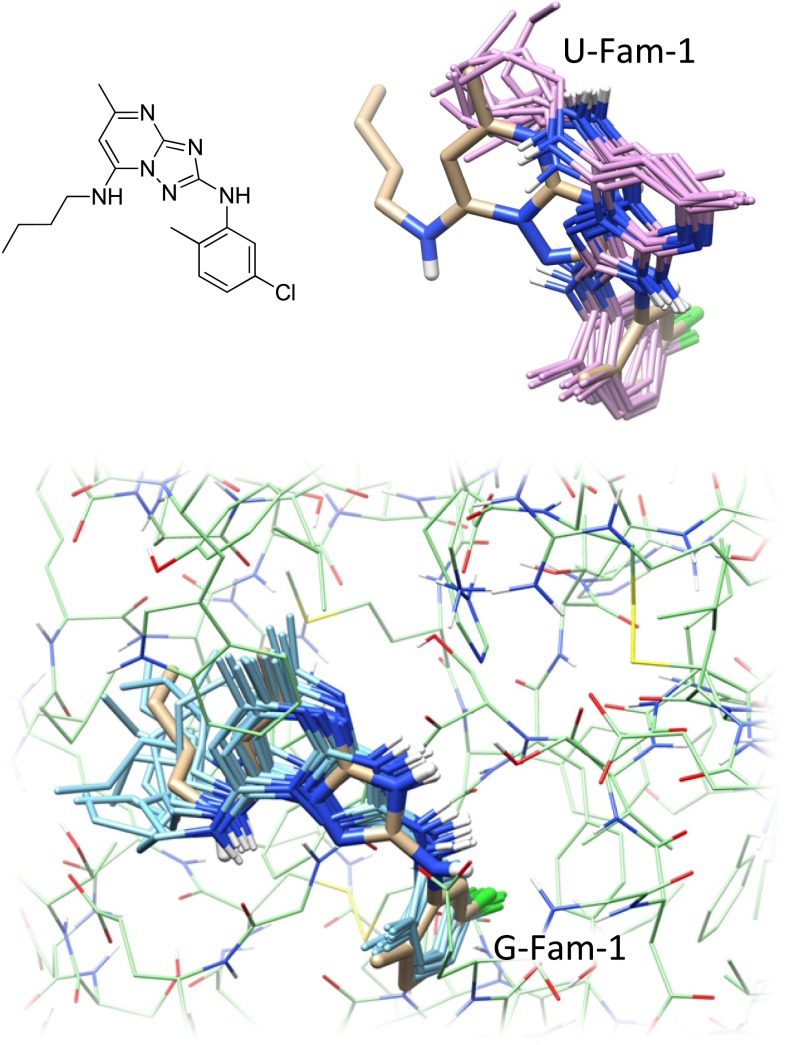
Prediction of the bound configuration of small-molecule ligands that differ substantially from the cognate ligand of a protein co-crystal structure is much more challenging than re-docking the cognate ligand. Success rates for cross-docking in the range of 20-30 % are common. We present an approach that uses structural information known prior to a particular cutoff-date to make predictions on ligands whose bounds structures were determined later. The knowledge-guided docking protocol was tested on a set of ten protein targets using a total of 949 ligands. The benchmark data set, called PINC ("PINC Is Not Cognate"), is publicly available. Protein pocket similarity was used to choose representative structures for ensemble-docking. The docking protocol made use of known ligand poses prior to the cutoff-date, both to help guide the configurational search and to adjust the rank of predicted poses. Overall, the top-scoring pose family was correct over 60 % of the time, with the top-two pose families approaching a 75 % success rate. Correct poses among all those predicted were identified nearly 90 % of the time. The largest improvements came from the use of molecular similarity to improve ligand pose rankings and the strategy for identifying representative protein structures. With the exception of a single outlier target, the knowledge-guided docking protocol produced results matching the quality of cognate-ligand re-docking, but it did so on a very challenging temporally-segregated cross-docking benchmark.
Figures
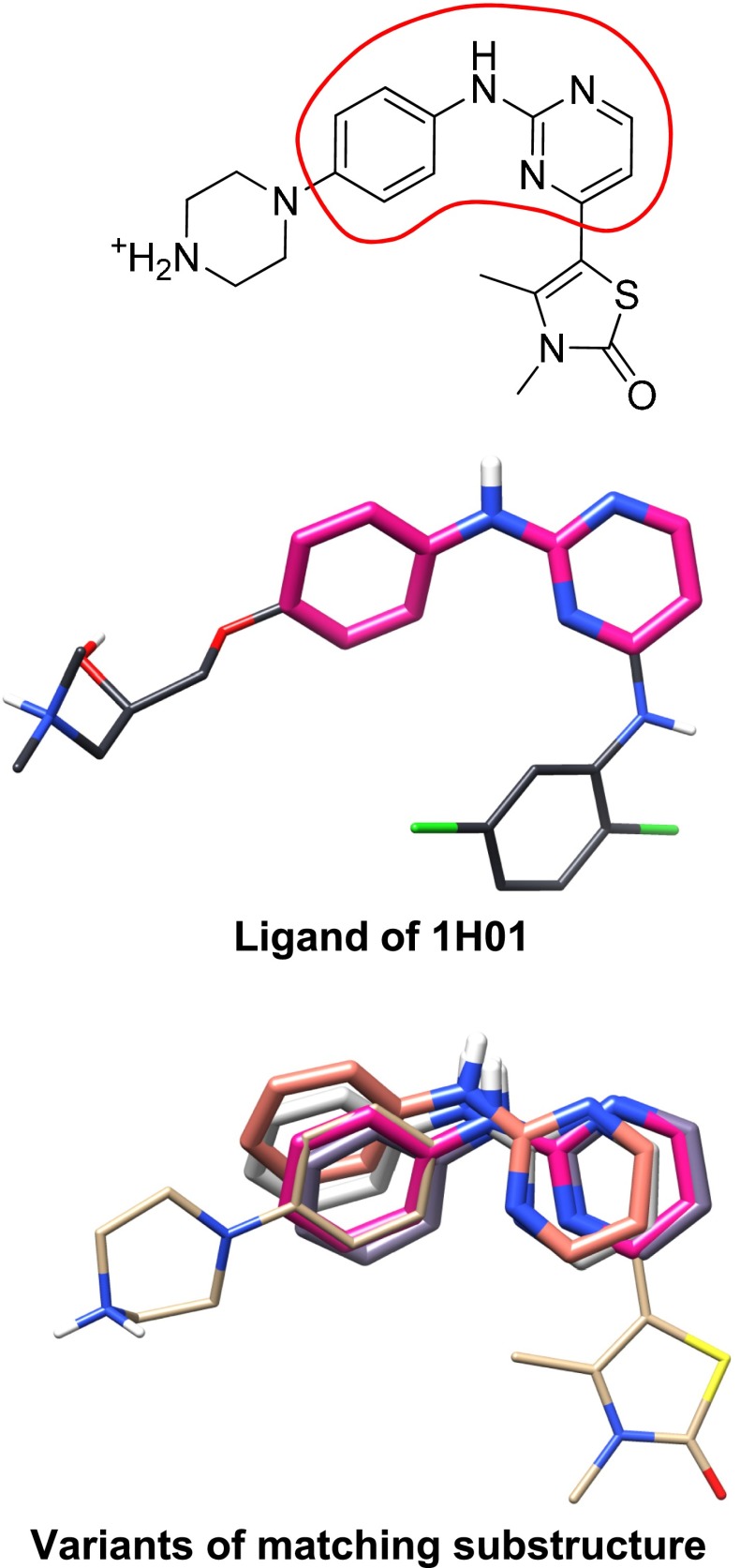
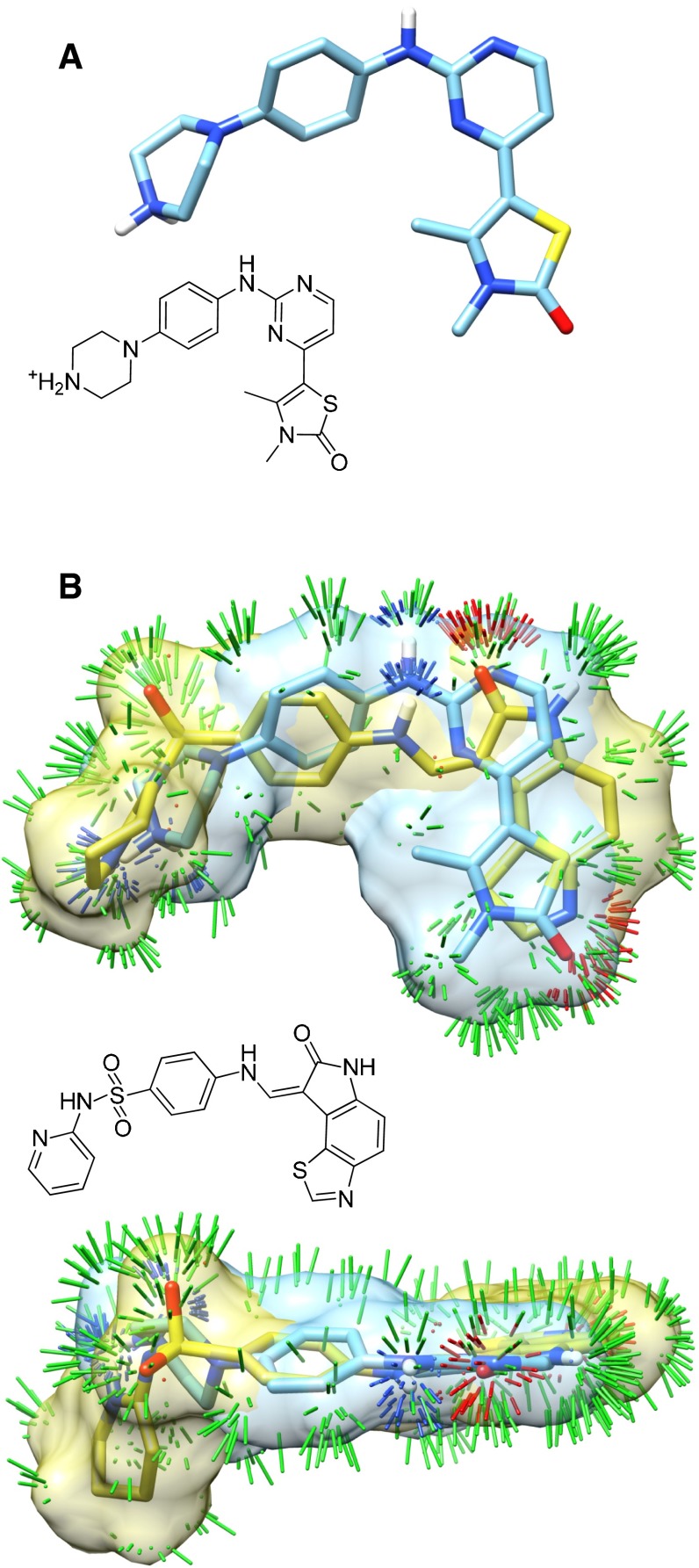
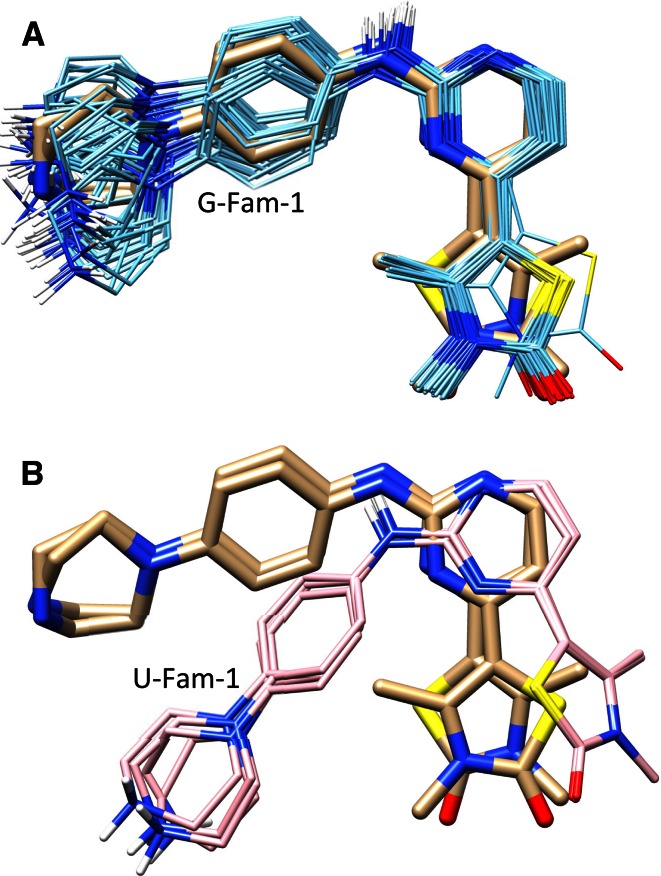
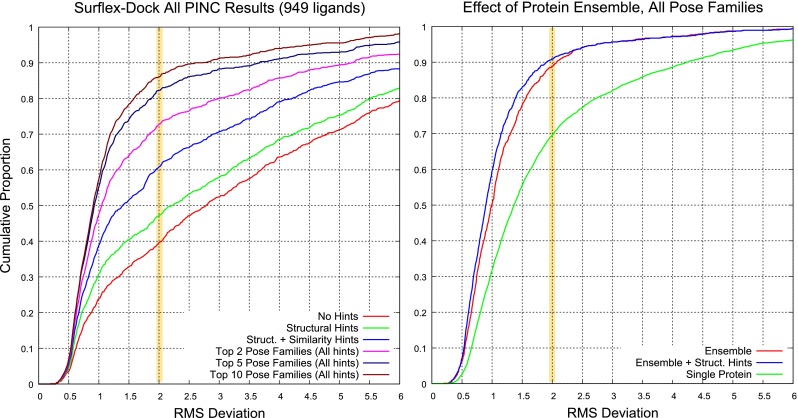
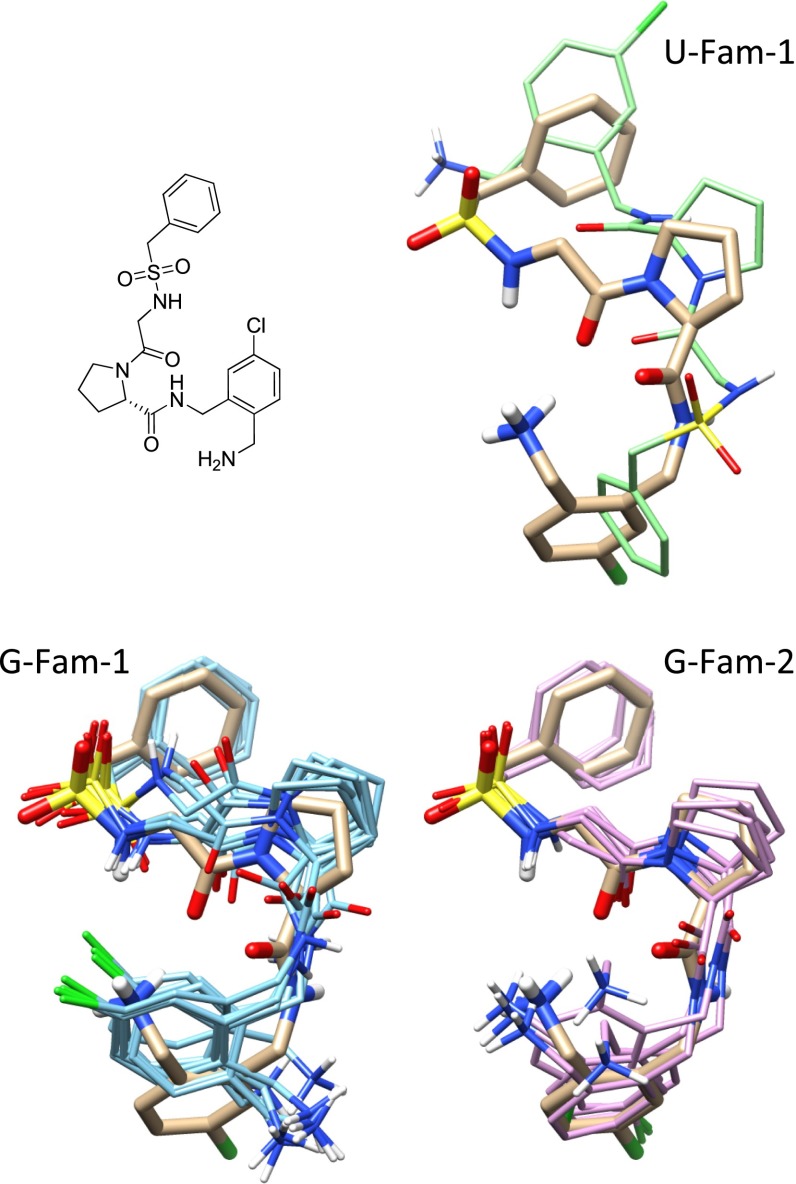
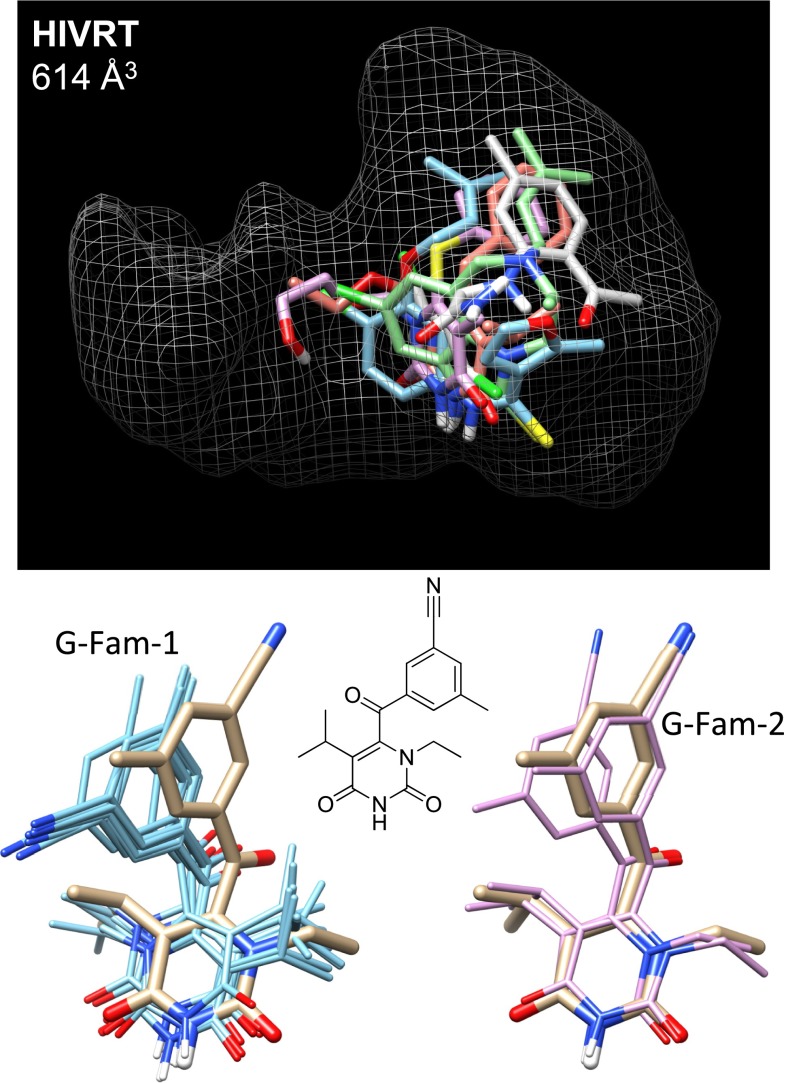
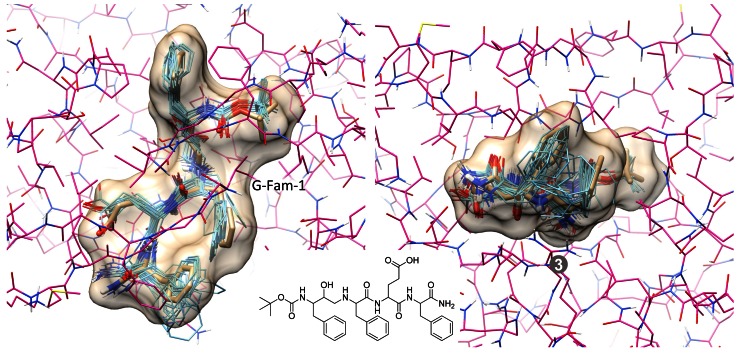
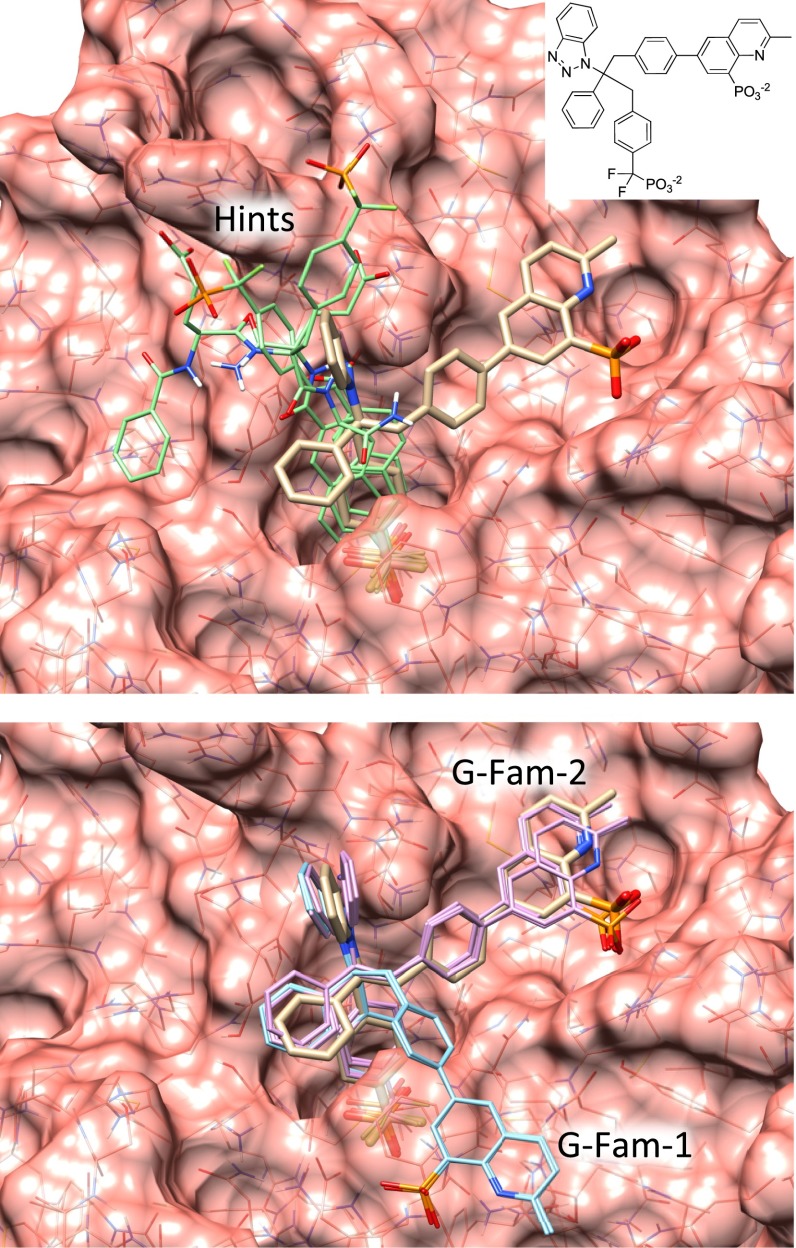
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
