

Group Selection and Contribution of Minority Variants during Virus Adaptation Determines Virus Fitness and Phenotype
- PMID: 25941809
- PMCID: PMC4420505
- DOI: 10.1371/journal.ppat.1004838
Group Selection and Contribution of Minority Variants during Virus Adaptation Determines Virus Fitness and Phenotype
Abstract
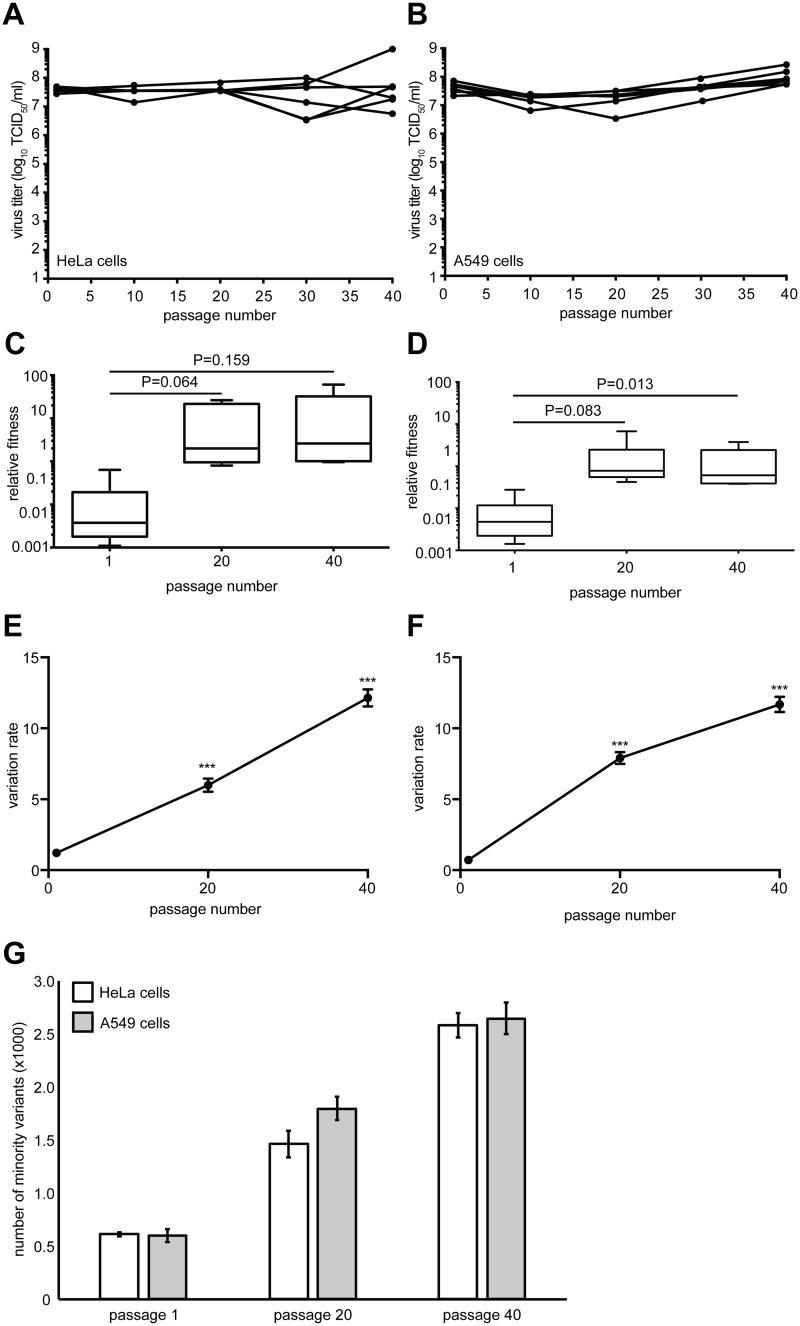
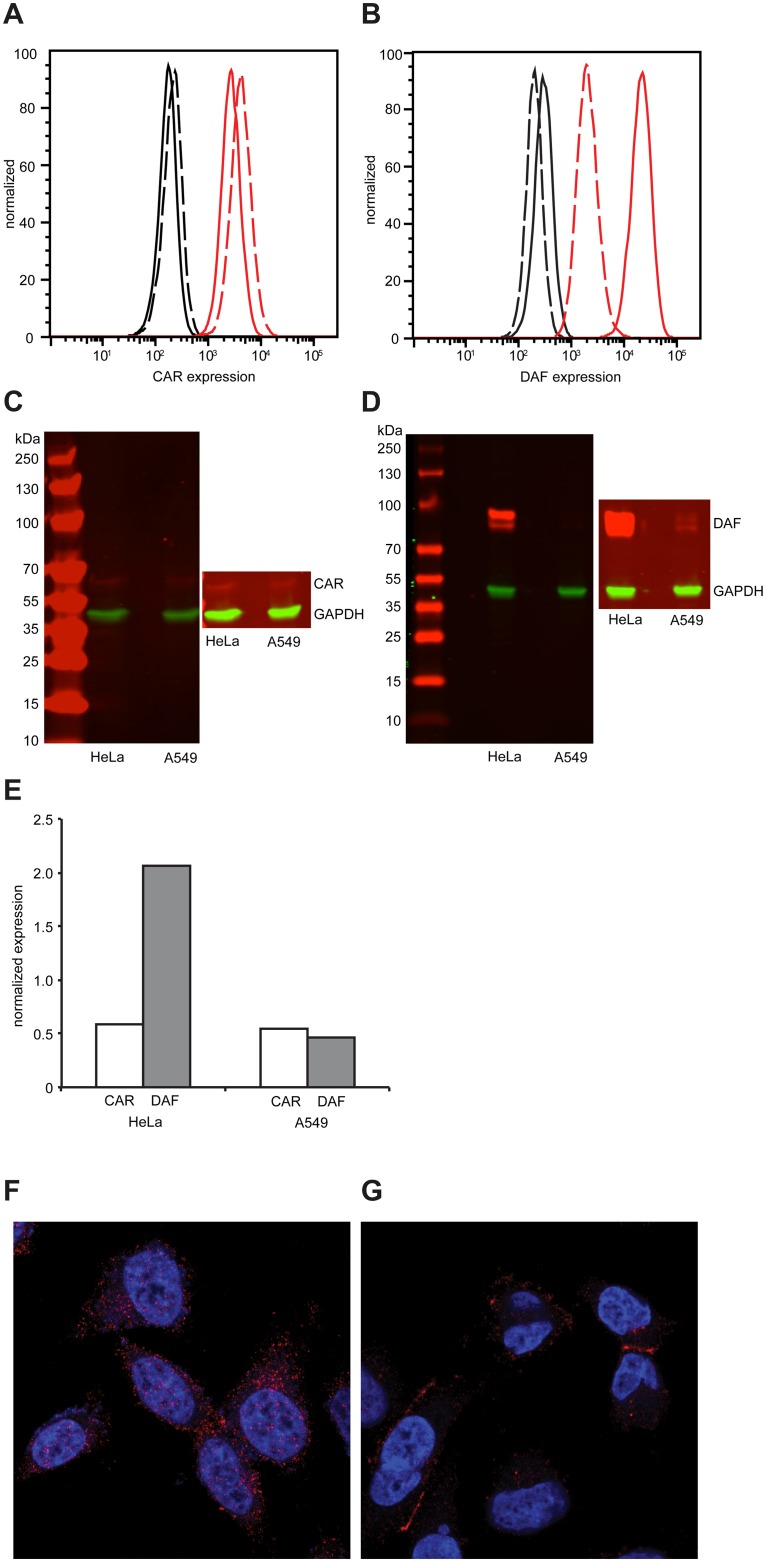
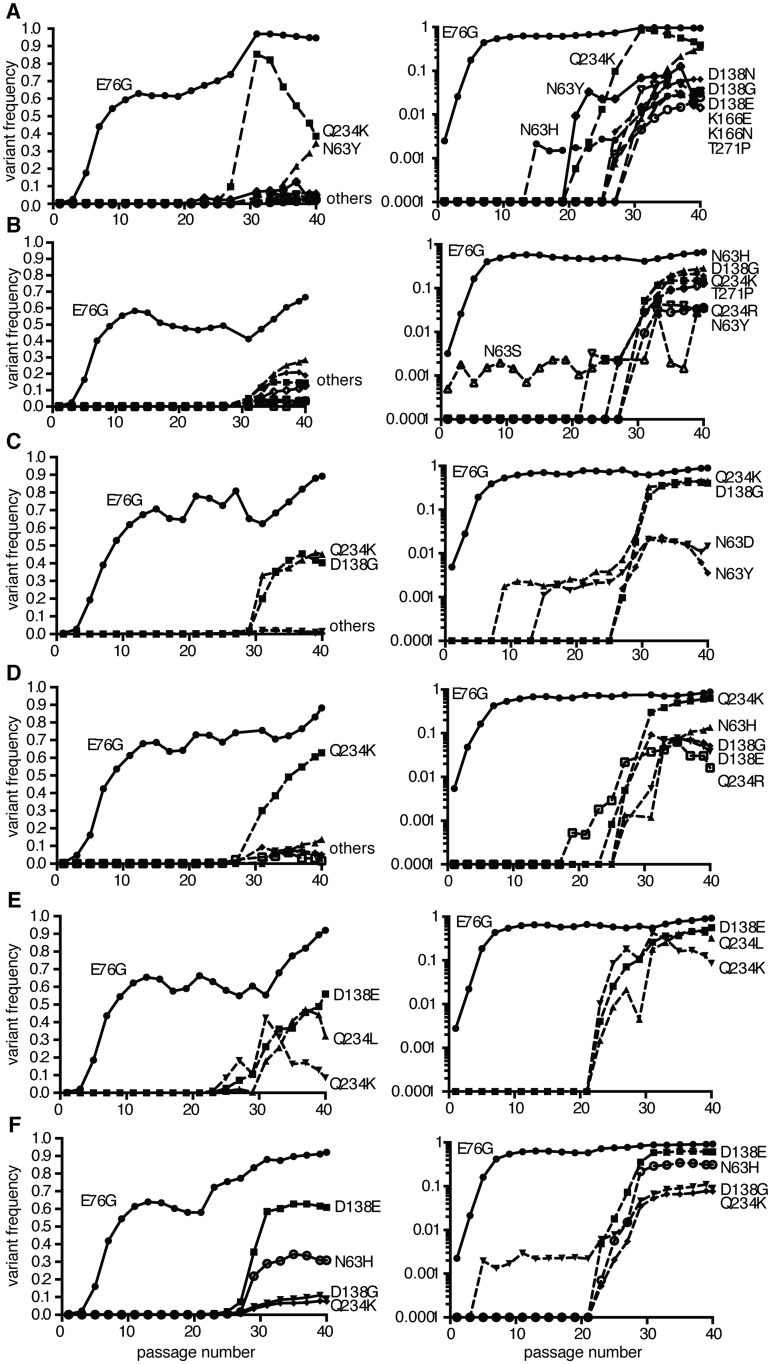
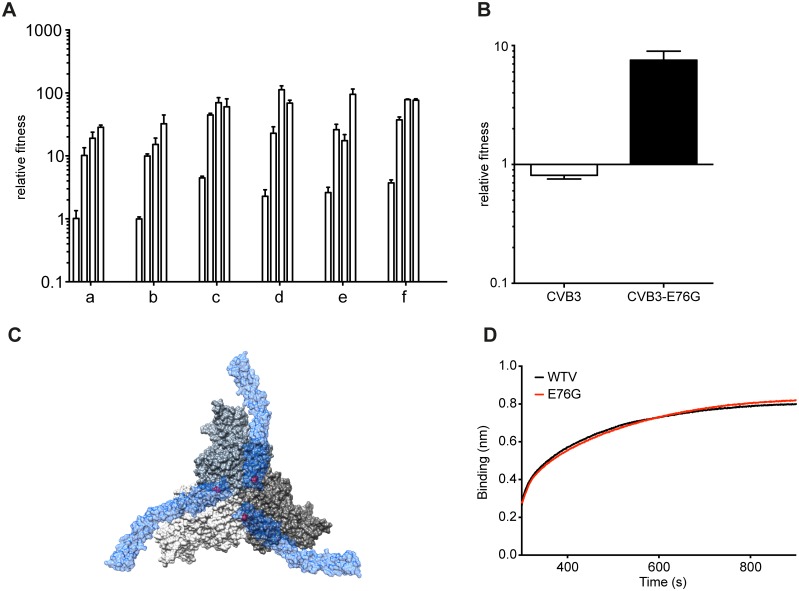
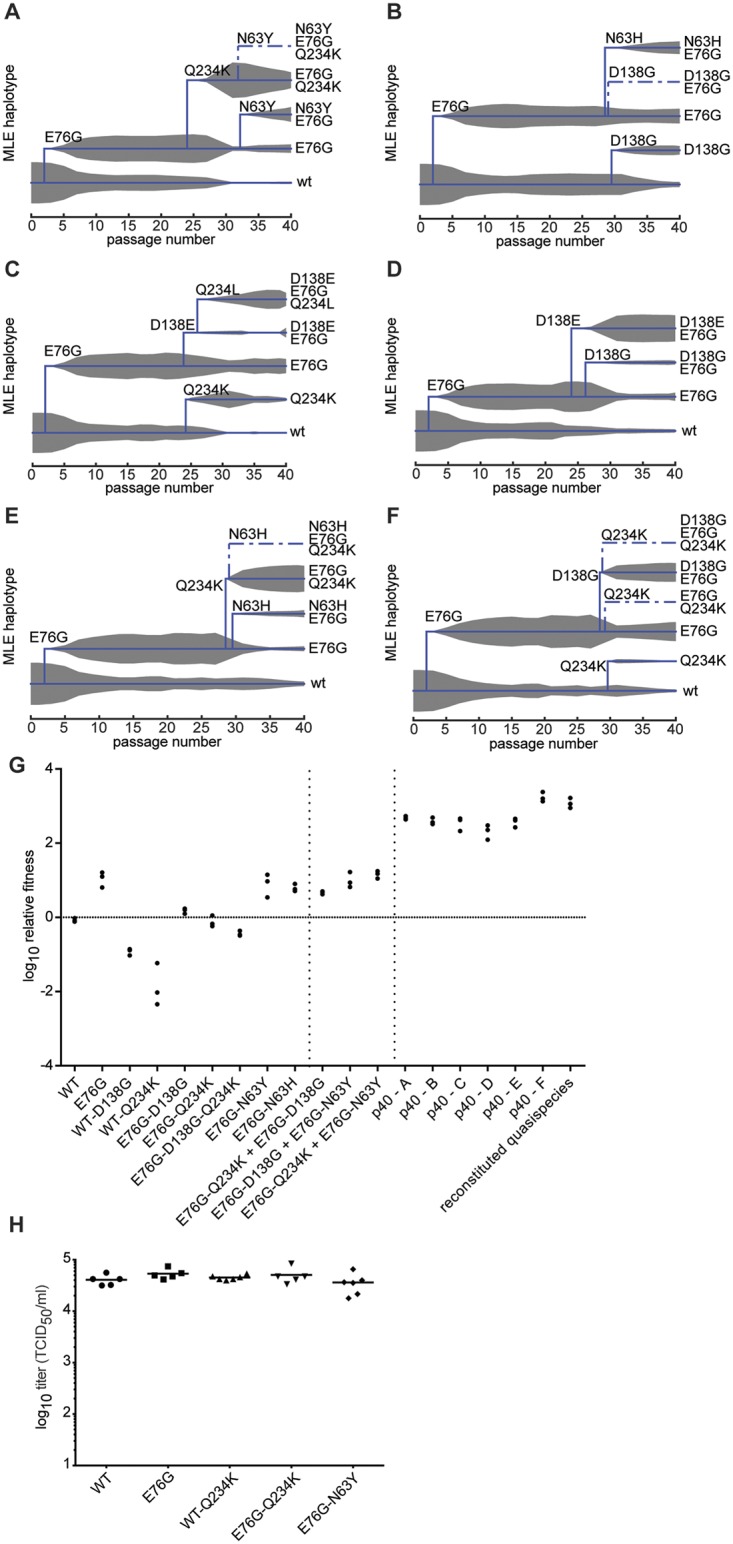
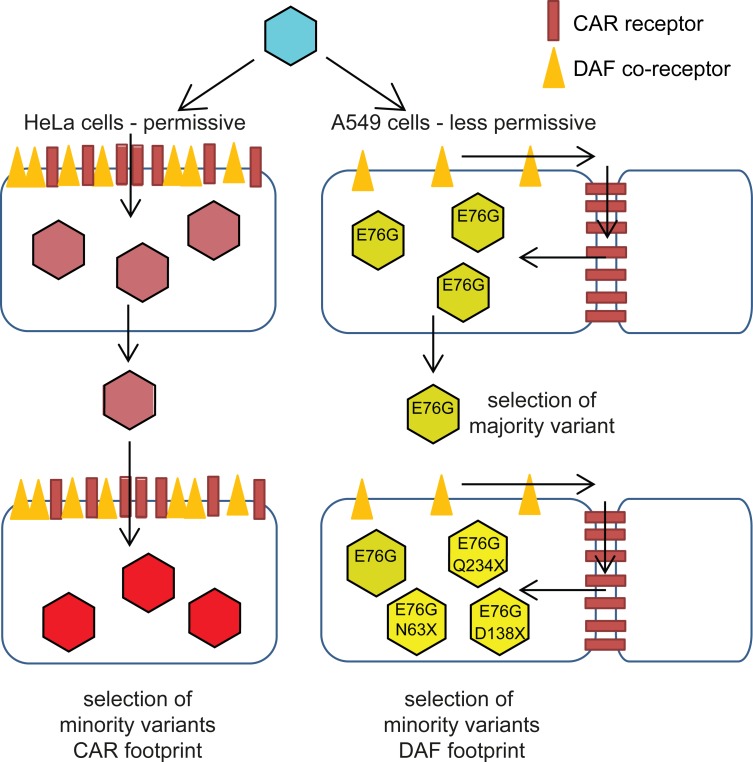
Understanding how a pathogen colonizes and adapts to a new host environment is a primary aim in studying emerging infectious diseases. Adaptive mutations arise among the thousands of variants generated during RNA virus infection, and identifying these variants will shed light onto how changes in tropism and species jumps can occur. Here, we adapted Coxsackie virus B3 to a highly permissive and less permissive environment. Using deep sequencing and bioinformatics, we identified a multi-step adaptive process to adaptation involving residues in the receptor footprints that correlated with receptor availability and with increase in virus fitness in an environment-specific manner. We show that adaptation occurs by selection of a dominant mutation followed by group selection of minority variants that together, confer the fitness increase observed in the population, rather than selection of a single dominant genotype.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
