

Expanding the clinical and molecular characteristics of PIGT-CDG, a disorder of glycosylphosphatidylinositol anchors
- PMID: 25943031
- PMCID: PMC6341466
- DOI: 10.1016/j.ymgme.2015.04.007
Expanding the clinical and molecular characteristics of PIGT-CDG, a disorder of glycosylphosphatidylinositol anchors
Abstract
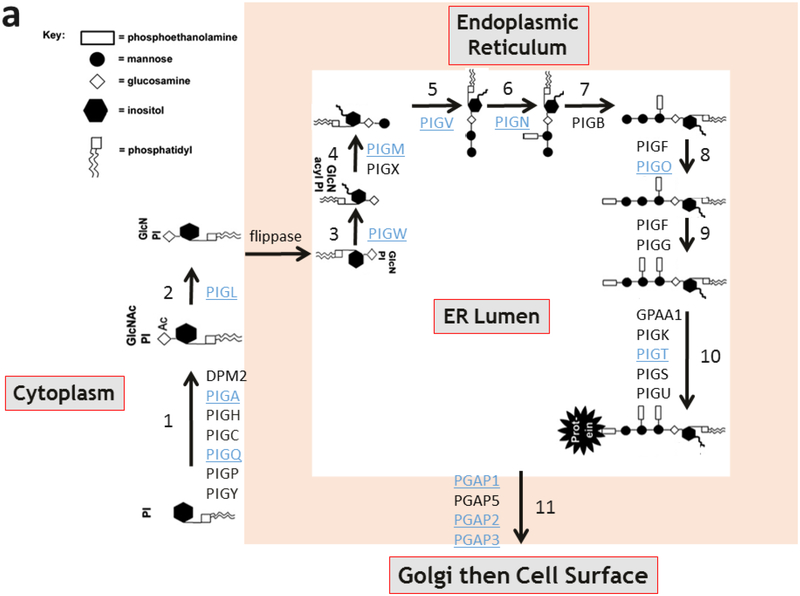
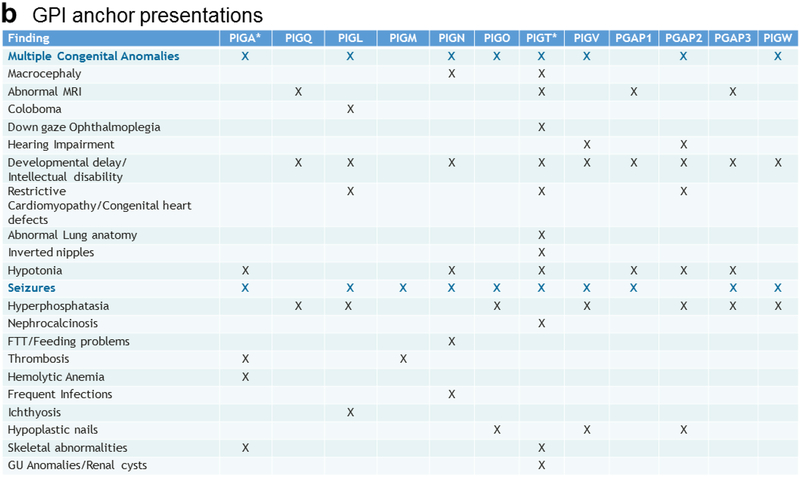
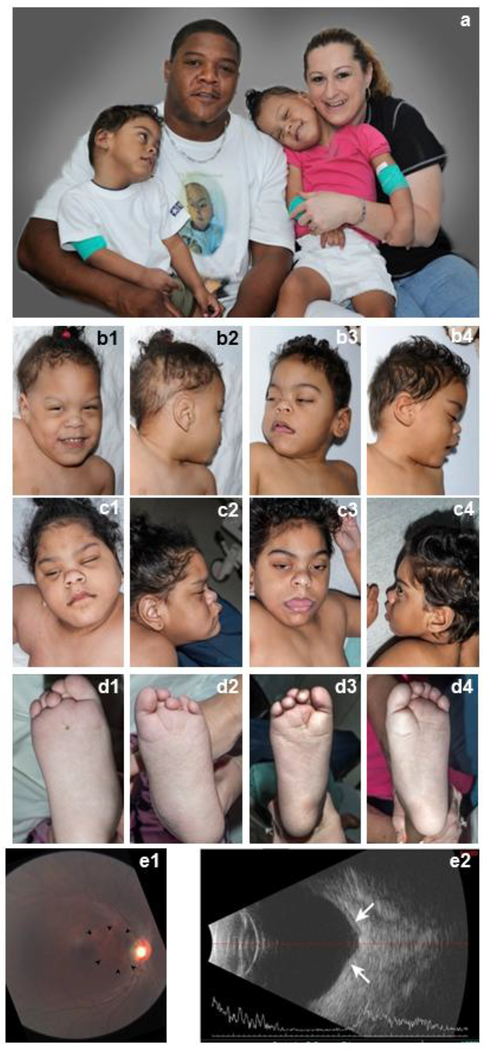
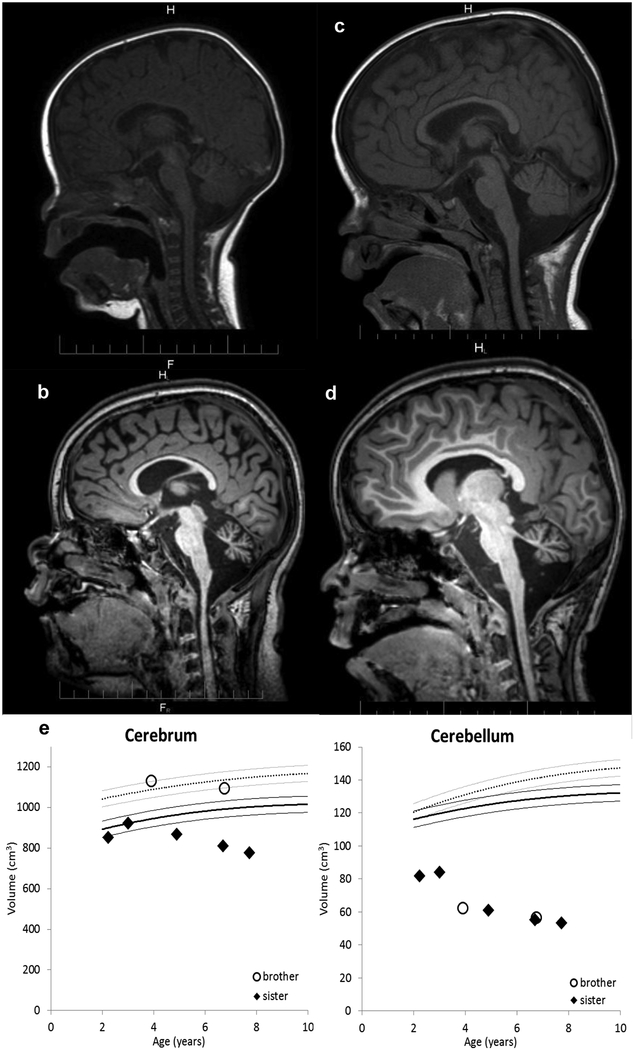
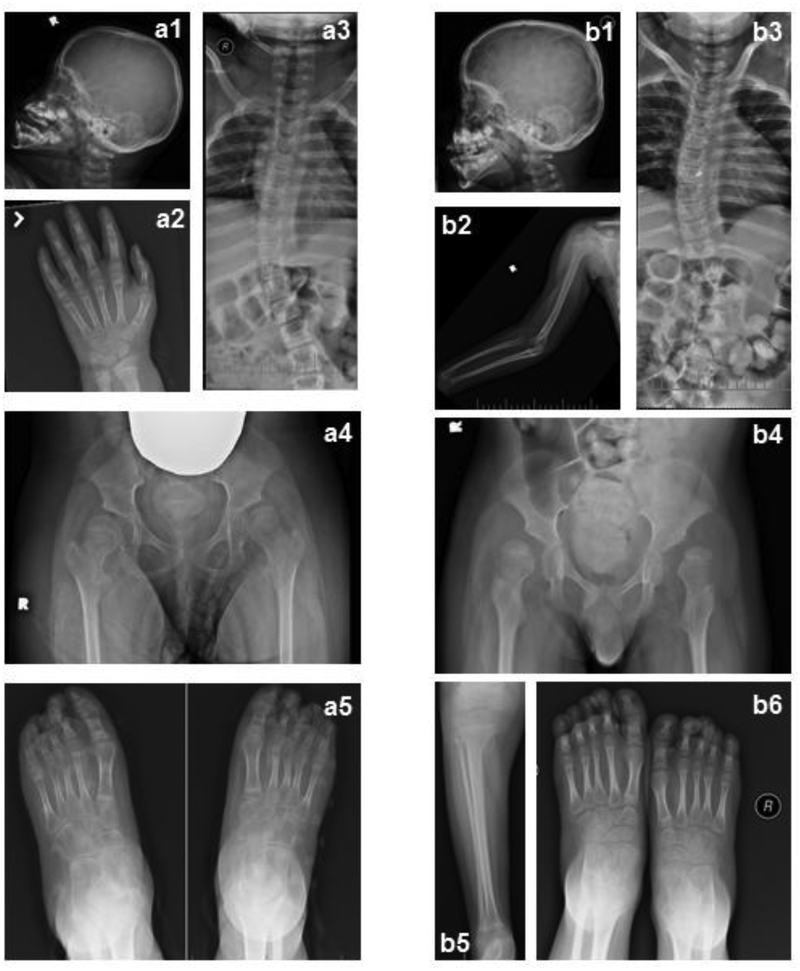
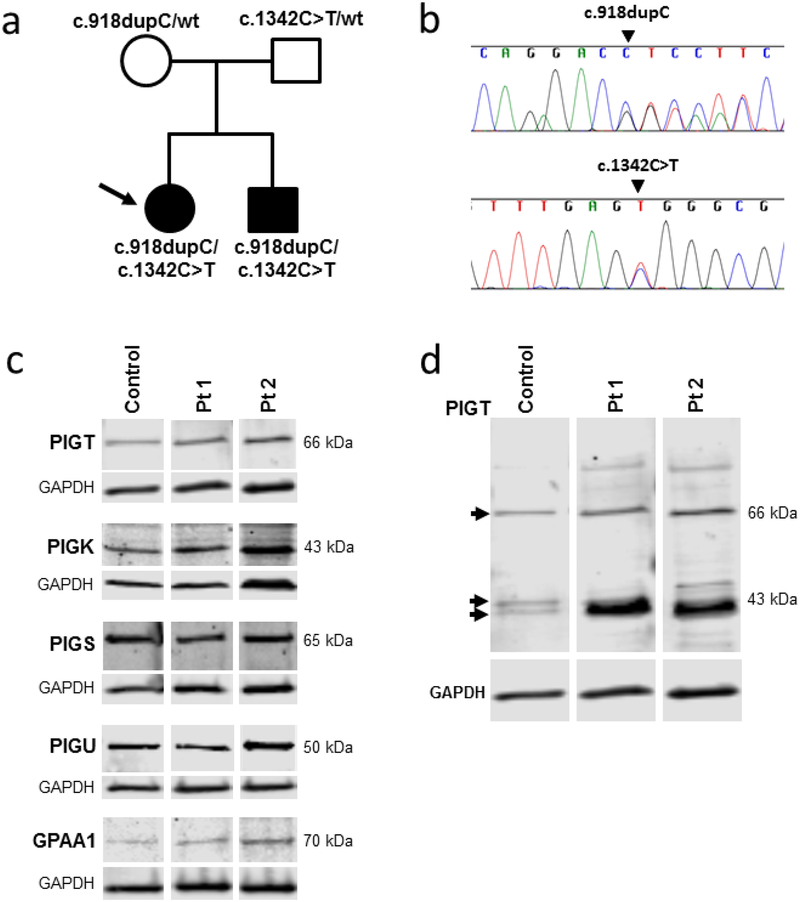
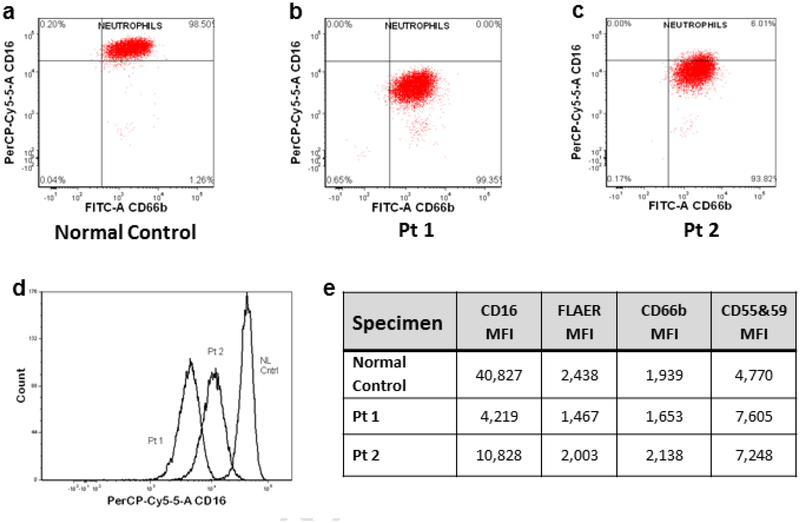
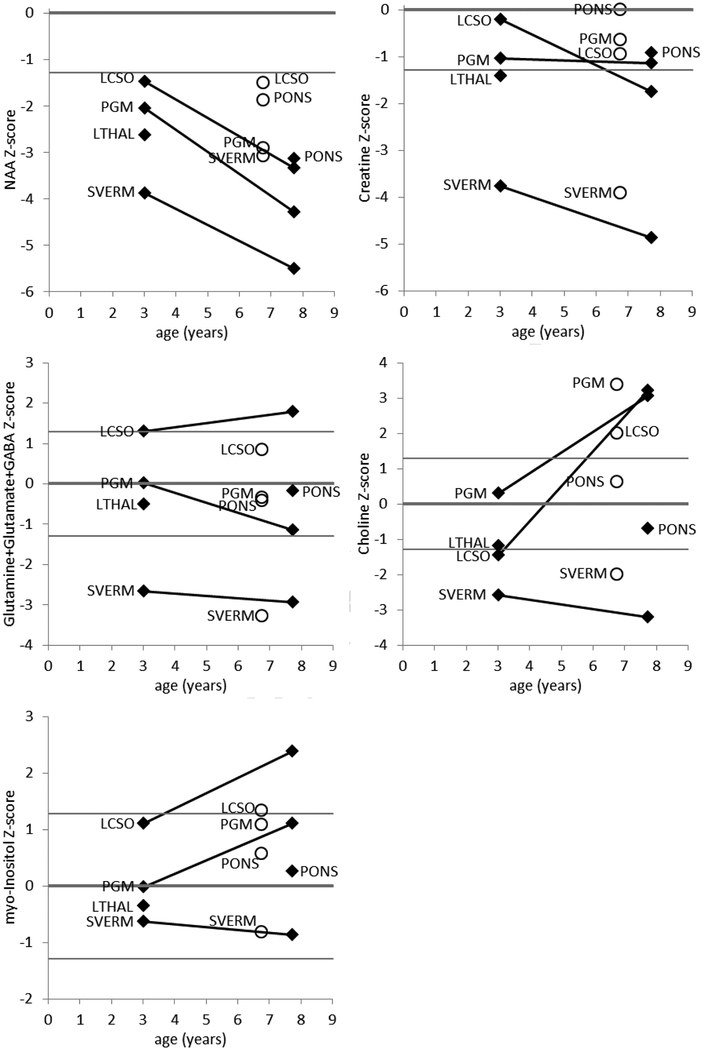
PIGT-CDG, an autosomal recessive syndromic intellectual disability disorder of glycosylphosphatidylinositol (GPI) anchors, was recently described in two independent kindreds [Multiple Congenital Anomalies-Hypotonia-Seizures Syndrome 3 (OMIM, #615398)]. PIGT encodes phosphatidylinositol-glycan biosynthesis class T, a subunit of the heteropentameric transamidase complex that facilitates the transfer of GPI to proteins. GPI facilitates attachment (anchoring) of proteins to cell membranes. We describe, at ages 7 and 6 years, two children of non-consanguineous parents; they had hypotonia, severe global developmental delay, and intractable seizures along with endocrine, ophthalmologic, skeletal, hearing, and cardiac anomalies. Exome sequencing revealed that both siblings had compound heterozygous variants in PIGT (NM_015937.5), i.e., c.918dupC, a novel duplication leading to a frameshift, and c.1342C > T encoding a previously described missense variant. Flow cytometry studies showed decreased surface expression of GPI-anchored proteins on granulocytes, consistent with findings in previous cases. These siblings further delineate the clinical spectrum of PIGT-CDG, reemphasize the neuro-ophthalmologic presentation, clarify the endocrine features, and add hypermobility, low CSF albumin quotient, and hearing loss to the phenotypic spectrum. Our results emphasize that GPI anchor-related congenital disorders of glycosylation (CDGs) should be considered in subjects with early onset severe seizure disorders and dysmorphic facial features, even in the presence of a normal carbohydrate-deficient transferrin pattern and N-glycan profiling. Currently available screening for CDGs will not reliably detect this family of disorders, and our case reaffirms that the use of flow cytometry and genetic testing is essential for diagnosis in this group of disorders.
Keywords: Congenital disorder of glycosylation; Exome; Flow cytometry; Glycosylphosphatidylinositol anchor; PIGT-CDG; Phenotype.
Copyright © 2015. Published by Elsevier Inc.
Conflict of interest statement
Conflict of interest
The authors declare no conflict of interest.
Figures
References
-
- Nosjean O, Briolay A, Roux B, Mammalian GPI proteins: sorting, membrane residence and functions, Biochim. Biophys. Acta 1331 (1997) 153–186. - PubMed
-
- Ferguson MAJ, Kinoshita T, Hart GW, Glycosylphosphatidylinositol Anchors, in: Varki A, Cummings RD, Esko JD, Freeze HH, Stanley P, Bertozzi CR, Hart GW, Etzler ME (Eds), Essentials of Glycobiology, 2nd edition, Cold Spring Harbor (NY), Cold Spring Harbor Laboratory Press, 2009, Chapter 11. - PubMed
-
- Kinoshita T, Fujita M, Maeda Y, Biosynthesis, remodelling and functions of mammalian GPI-anchored proteins: recent progress, J. Biochem 144 (2008) 287–294. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
