

Apoptotic effects of high-dose rapamycin occur in S-phase of the cell cycle
- PMID: 25945415
- PMCID: PMC4614271
- DOI: 10.1080/15384101.2015.1046653
Apoptotic effects of high-dose rapamycin occur in S-phase of the cell cycle
Abstract
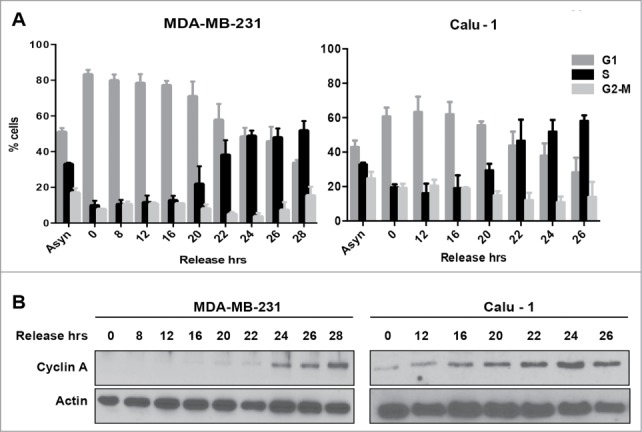
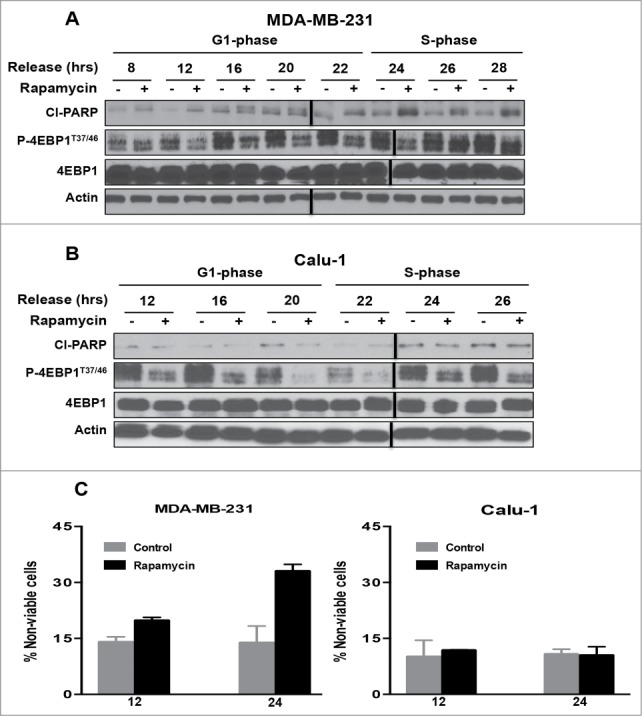
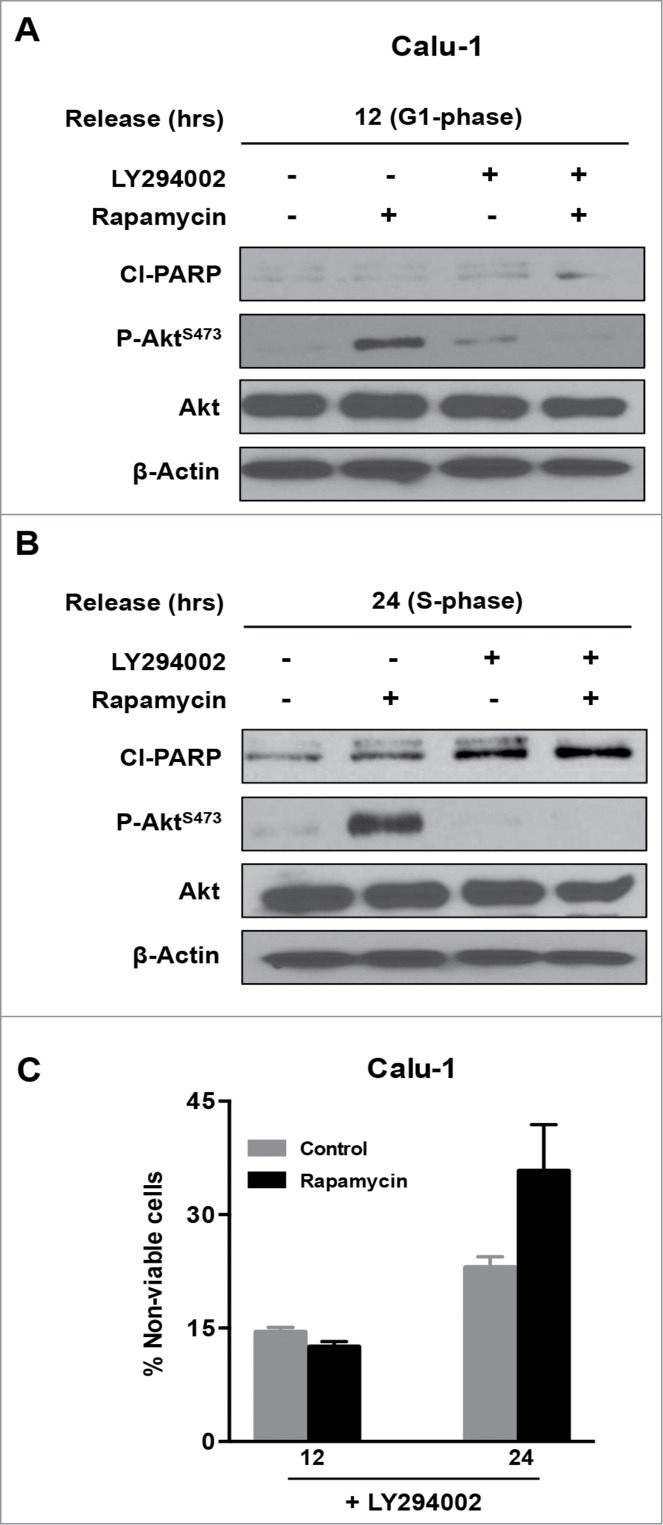
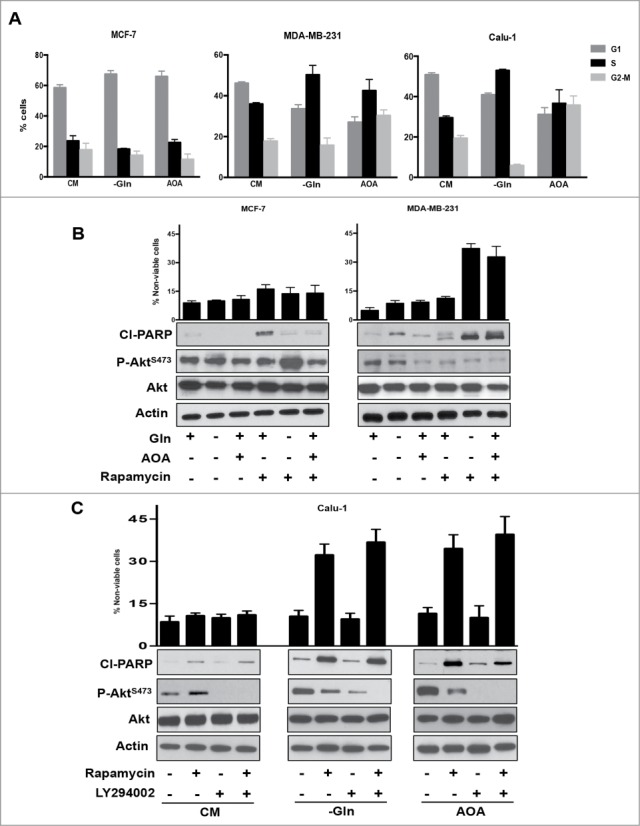
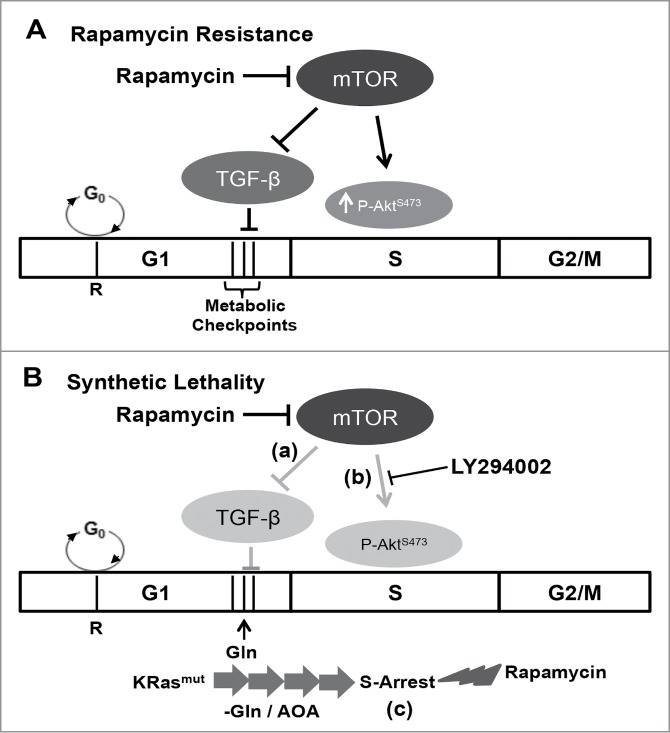
Mutations in genes encoding regulators of mTOR, the mammalian target of rapamycin, commonly provide survival signals in cancer cells. Rapamycin and analogs of rapamycin have been used with limited success in clinical trials to target mTOR-dependent survival signals in a variety of human cancers. Suppression of mTOR predominantly causes G1 cell cycle arrest, which likely contributes to the ineffectiveness of rapamycin-based therapeutic strategies. While rapamycin causes the accumulation of cells in G1, its effect in other cell cycle phases remains largely unexplored. We report here that when synchronized MDA-MB-231 breast cancer cells are allowed to progress into S-phase from G1, rapamycin activates the apoptotic machinery with a concomitant increase in cell death. In Calu-1 lung cancer cells, rapamycin induced a feedback increase in Akt phosphorylation at Ser473 in S-phase that mitigated rapamycin-induced apoptosis. However, sensitivity to rapamycin in S-phase could be reestablished if Akt phosphorylation was suppressed. We recently reported that glutamine (Gln) deprivation causes K-Ras mutant cancer cells to aberrantly arrest primarily in S-phase. Consistent with observed sensitivity of S-phase cells to rapamycin, interfering with Gln utilization sensitized both MDA-MB-231 and Calu-1 K-Ras mutant cancer cells to the apoptotic effect of rapamycin. Importantly, rapamycin induced substantially higher levels of cell death upon Gln depletion than that observed in cancer cells that were allowed to progress through S-phase after being synchronized in G1. We postulate that exploiting metabolic vulnerabilities in cancer cells such as S-phase arrest observed with K-Ras-driven cancer cells deprived of Gln, could be of great therapeutic potential.
Keywords: 4E-BP1, eIF4E binding protein-1; GOT, glutamate-oxaloacetate-transaminase; Gln, glutamine; PARP, poly-ADP-ribose polymerase; PI3K, phosphatidylinositol-3-kinase; S6K, S6 kinase; TGF-β, transforming growth factor-β.; cell cycle; eIF4E, eukaryotic initiation factor 4E; glutamine; mTOR; mTOR, mammalian target of rapamycin; mTORC1/2, mTOR complex 1/2; rapamycin; synthetic lethality.
Figures
Comment in
-
Cell cycle status dictates effectiveness of rapamycin.Cell Cycle. 2015;14(16):2556-7. doi: 10.1080/15384101.2015.1060778. Epub 2015 Jun 30. Cell Cycle. 2015. PMID: 26126171 Free PMC article. No abstract available.
References
-
- Fingar DC, Blenis J. Target of rapamycin (TOR): an integrator of nutrient and growth factor signals and coordinator of cell growth and cell cycle progression. Oncogene 2004; 23:3151-71; PMID:15094765; http://dx.doi.org/ 10.1038/sj.onc.1207542 - DOI - PubMed
-
- Ward PS, Thompson CB. Metabolic reprogramming: a cancer hallmark even warburg did not anticipate. Cancer Cell 2012; 21:297-308; PMID:22439925; http://dx.doi.org/ 10.1016/j.ccr.2012.02.014 - DOI - PMC - PubMed
-
- Foster DA. Phosphatidic acid signaling to mTOR: signals for the survival of human cancer cells. Biochim Biophys Acta 2009; 1791:949-55; PMID:19264150; http://dx.doi.org/ 10.1016/j.bbalip.2009.02.009 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous