

The solvent component of macromolecular crystals
- PMID: 25945568
- PMCID: PMC4427195
- DOI: 10.1107/S1399004715006045
The solvent component of macromolecular crystals
Abstract
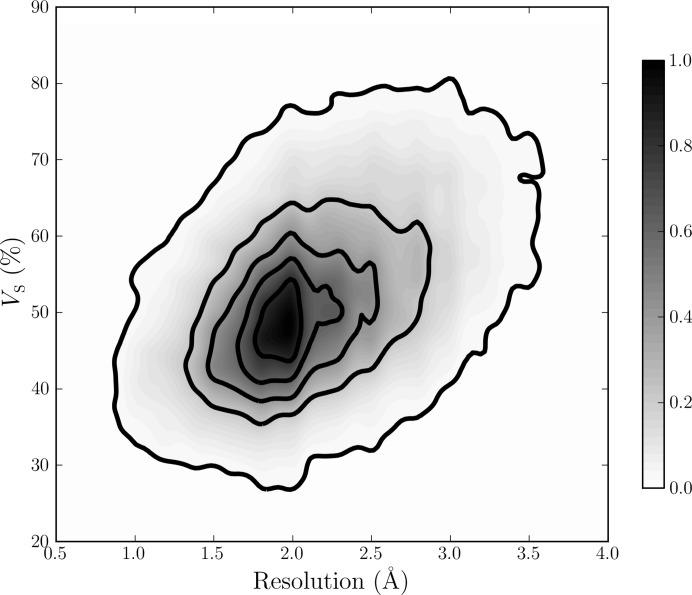
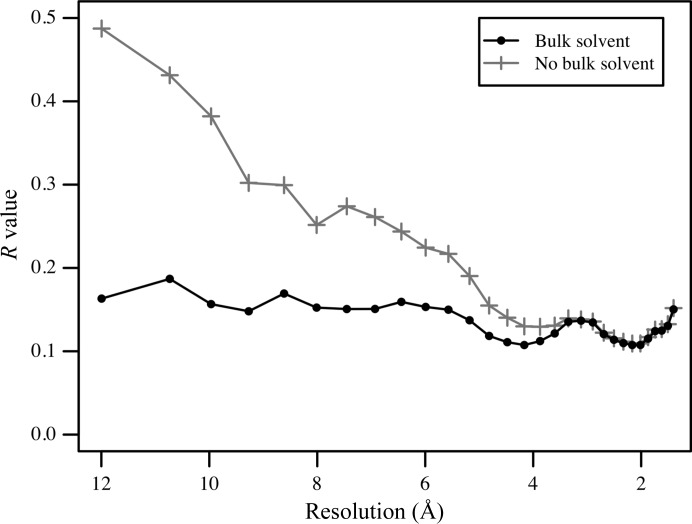
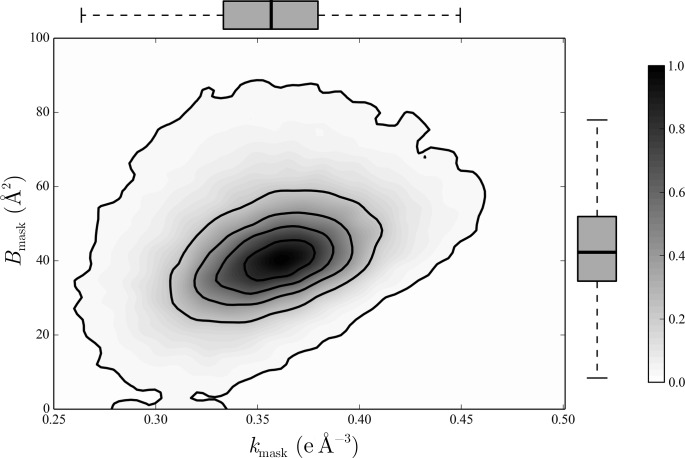
The mother liquor from which a biomolecular crystal is grown will contain water, buffer molecules, native ligands and cofactors, crystallization precipitants and additives, various metal ions, and often small-molecule ligands or inhibitors. On average, about half the volume of a biomolecular crystal consists of this mother liquor, whose components form the disordered bulk solvent. Its scattering contributions can be exploited in initial phasing and must be included in crystal structure refinement as a bulk-solvent model. Concomitantly, distinct electron density originating from ordered solvent components must be correctly identified and represented as part of the atomic crystal structure model. Herein, are reviewed (i) probabilistic bulk-solvent content estimates, (ii) the use of bulk-solvent density modification in phase improvement, (iii) bulk-solvent models and refinement of bulk-solvent contributions and (iv) modelling and validation of ordered solvent constituents. A brief summary is provided of current tools for bulk-solvent analysis and refinement, as well as of modelling, refinement and analysis of ordered solvent components, including small-molecule ligands.
Keywords: bulk solvent; macromolecular crystals; ordered solvent; solvent content.
Figures
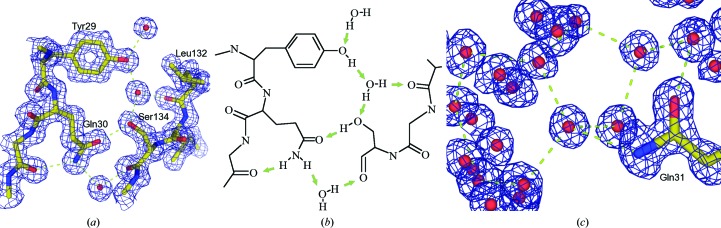
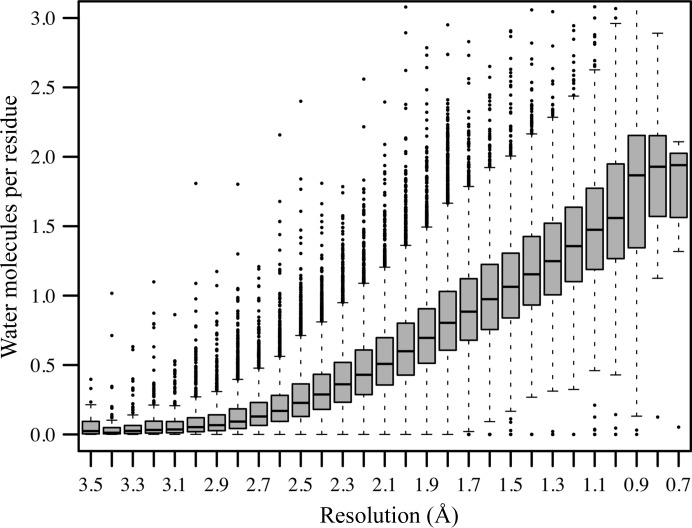
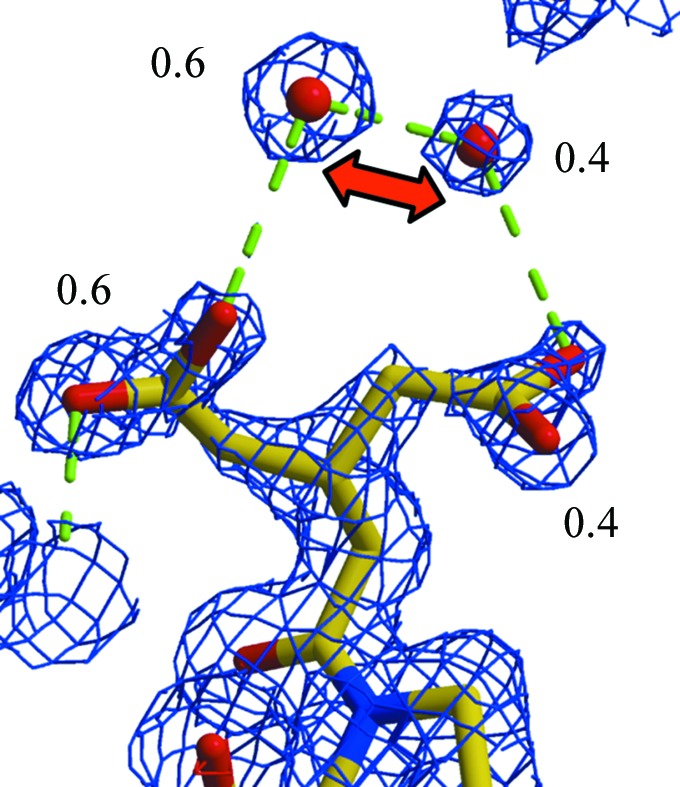
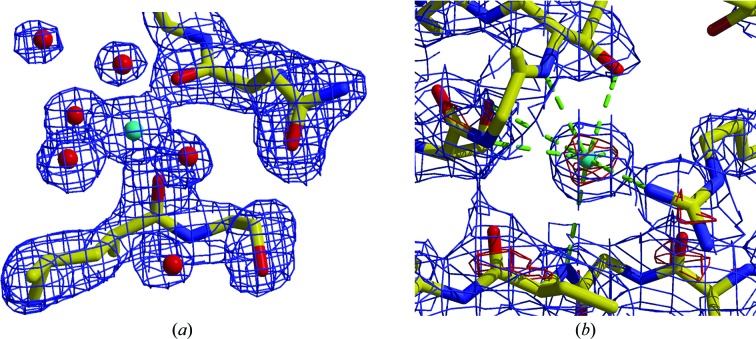
Similar articles
-
Prediction of models for ordered solvent in macromolecular structures by a classifier based upon resolution-independent projections of local feature data.Acta Crystallogr D Struct Biol. 2019 Aug 1;75(Pt 8):696-717. doi: 10.1107/S2059798319008933. Epub 2019 Jul 30. Acta Crystallogr D Struct Biol. 2019. PMID: 31373570 Free PMC article.
-
Accounting for nonuniformity of bulk-solvent: A mosaic model.Protein Sci. 2024 Mar;33(3):e4909. doi: 10.1002/pro.4909. Protein Sci. 2024. PMID: 38358136 Free PMC article.
-
Integral equation models for solvent in macromolecular crystals.J Chem Phys. 2022 Jan 7;156(1):014801. doi: 10.1063/5.0070869. J Chem Phys. 2022. PMID: 34998331 Free PMC article.
-
Optimization of crystallization conditions for biological macromolecules.Acta Crystallogr F Struct Biol Commun. 2014 Nov;70(Pt 11):1445-67. doi: 10.1107/S2053230X14019670. Epub 2014 Oct 31. Acta Crystallogr F Struct Biol Commun. 2014. PMID: 25372810 Free PMC article. Review.
-
So how do you know you have a macromolecular complex?Acta Crystallogr D Biol Crystallogr. 2007 Jan;63(Pt 1):17-25. doi: 10.1107/S0907444906047044. Epub 2006 Dec 13. Acta Crystallogr D Biol Crystallogr. 2007. PMID: 17164522 Free PMC article. Review.
Cited by
-
Nucleobase carbonyl groups are poor Mg2+ inner-sphere binders but excellent monovalent ion binders-a critical PDB survey.RNA. 2019 Feb;25(2):173-192. doi: 10.1261/rna.068437.118. Epub 2018 Nov 8. RNA. 2019. PMID: 30409785 Free PMC article.
-
Mg2+ ions: do they bind to nucleobase nitrogens?Nucleic Acids Res. 2017 Jan 25;45(2):987-1004. doi: 10.1093/nar/gkw1175. Epub 2016 Dec 6. Nucleic Acids Res. 2017. PMID: 27923930 Free PMC article.
-
Evaluation of models determined by neutron diffraction and proposed improvements to their validation and deposition.Acta Crystallogr D Struct Biol. 2018 Aug 1;74(Pt 8):800-813. doi: 10.1107/S2059798318004588. Epub 2018 Jul 24. Acta Crystallogr D Struct Biol. 2018. PMID: 30082516 Free PMC article. Review.
-
What are the current limits on determination of protonation state using neutron macromolecular crystallography?Methods Enzymol. 2020;634:225-255. doi: 10.1016/bs.mie.2020.01.008. Epub 2020 Feb 13. Methods Enzymol. 2020. PMID: 32093835 Free PMC article.
-
The T2 structure of polycrystalline cubic human insulin.Acta Crystallogr D Struct Biol. 2023 May 1;79(Pt 5):374-386. doi: 10.1107/S2059798323001328. Epub 2023 Apr 11. Acta Crystallogr D Struct Biol. 2023. PMID: 37039669 Free PMC article.
References
-
- Abrahams, J. P. & Leslie, A. G. W. (1996). Acta Cryst. D52, 30–42. - PubMed
-
- Adams, P. D. et al. (2010). Acta Cryst. D66, 213–221.
-
- Afonine, P. V. & Adams, P. D. (2012). Comput. Crystallogr. Newsl. 3, 18–21.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
