

Machine learning applications in genetics and genomics
- PMID: 25948244
- PMCID: PMC5204302
- DOI: 10.1038/nrg3920
Machine learning applications in genetics and genomics
Abstract
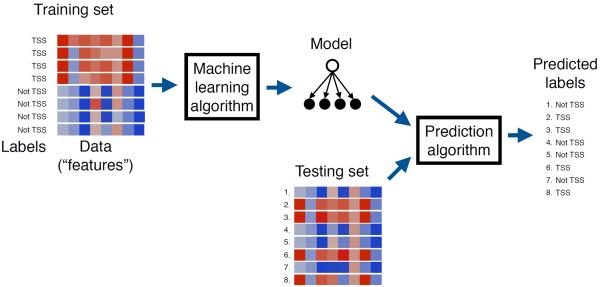
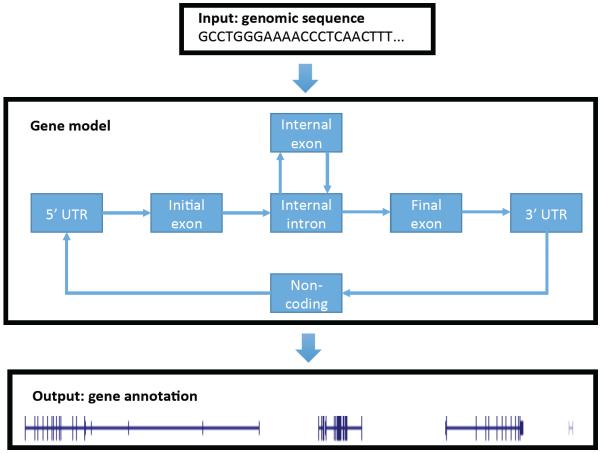
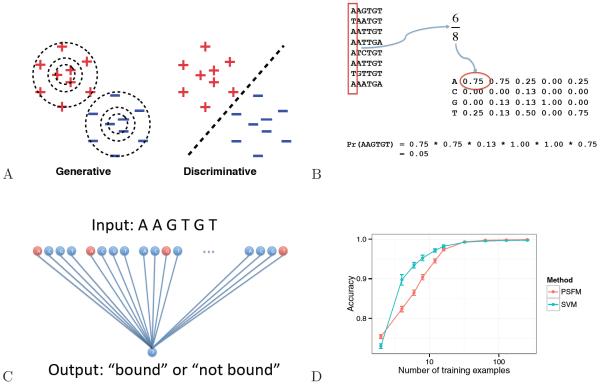
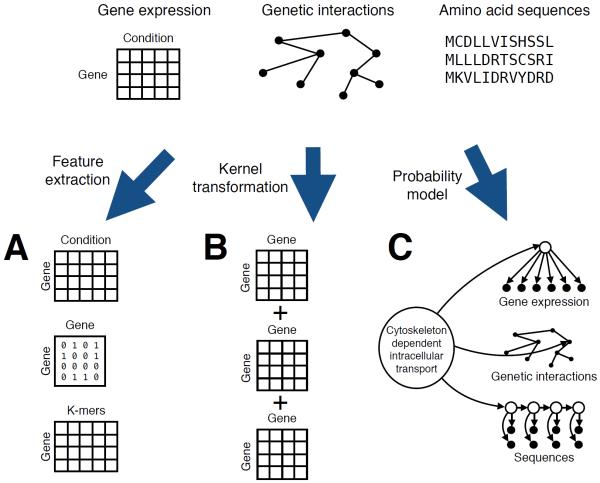
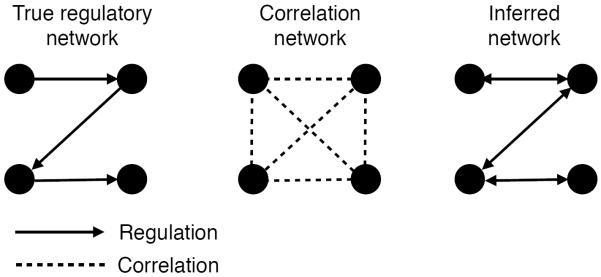
The field of machine learning, which aims to develop computer algorithms that improve with experience, holds promise to enable computers to assist humans in the analysis of large, complex data sets. Here, we provide an overview of machine learning applications for the analysis of genome sequencing data sets, including the annotation of sequence elements and epigenetic, proteomic or metabolomic data. We present considerations and recurrent challenges in the application of supervised, semi-supervised and unsupervised machine learning methods, as well as of generative and discriminative modelling approaches. We provide general guidelines to assist in the selection of these machine learning methods and their practical application for the analysis of genetic and genomic data sets.
Figures

References
-
- Mitchell T. Machine Learning. McGraw-Hill; 1997.
-
- Degroeve S, Baets BD, de Peer YV, Rouz P. Feature subset selection for splice site prediction. Bioinformatics. 2002;18:S75–S83. - PubMed
-
- Bucher P. Weight matrix description of four eukaryotic RNA polymerase II promoter elements derived from 502 unrelated promoter sequences. Journal of Molecular Biology. 1990;4:563–578. - PubMed
-
- Heintzman N, et al. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nature Genetics. 2007;39:311–318. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
