

Editorial
doi: 10.1161/CIRCRESAHA.115.306439.
Adrenergic and cholinergic plasticity in heart failure
Affiliations
- PMID: 25953921
- PMCID: PMC4428611
- DOI: 10.1161/CIRCRESAHA.115.306439
Item in Clipboard
Editorial
Adrenergic and cholinergic plasticity in heart failure
Circ Res.
.
No abstract available
Keywords: Editorials; adrenergic agents; heart failure; muscarinic agents; sudden death.
Figures
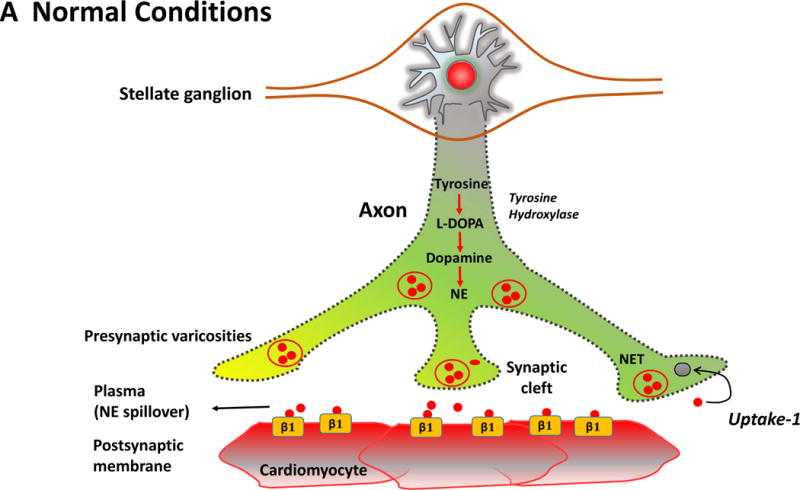
The Figure summarizes the plasticity of neurotransmitters in the sympathetic
nerve ganglion in conjunction with postsynaptic receptors on cardiac myocytes.
In the normal heart (Panel A), myocardial sympathetic nerves originating in the
stellate ganglion secrete norepinephrine (NE, red) to increase heart rate and
myocardial contractility through β1-adrenergic receptor signaling. The
interstitial NE level at the receptor is dependent upon release as well as the
presynaptic reuptake (Uptake 1) mechanism for recycling it into the presynaptic
terminal. Parasympathetic control of ventricular contractility (not illustrated)
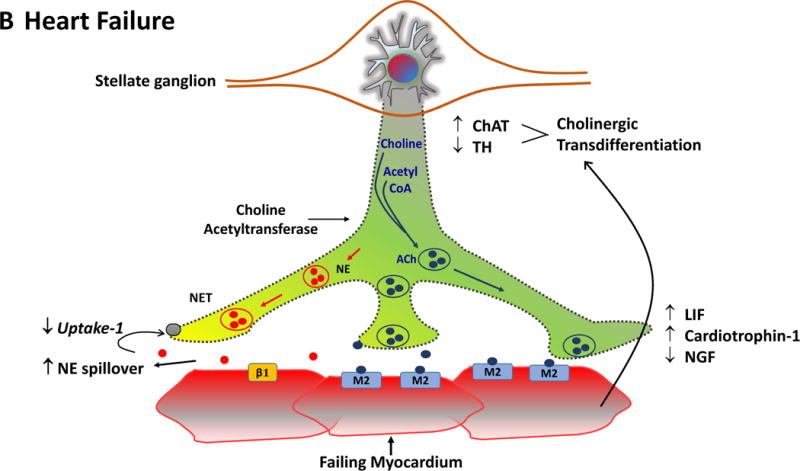
is generally felt to play a minor role. In heart failure (Panel B), plasticity
of neurons in the sympathetic ganglia leads to transdifferentiation of some cell
bodies to a cholinergic phenotype (blue). Some sympathetic nerves now express
Choline Acetyltransferase (AChAT) and tyrosine hydroxylase (TH) expression
decreases. This transdifferentiation is promoted by Leukemia Inhibitory Factor
(LIF), Cardiotrophin-1 expression and Nerve Growth Factor (NGF) released from
the myocardium. Reductions in NE reuptake as well as increased circulating NE
levels elevate interstitial NE and lead to post-synaptic down regulation of
β1 receptor density, uncoupling of β1 receptors with G proteins
and desensitization of β adrenergic signaling. In heart failure,
cardiomyocytes paradoxically increase the expression of muscarinic (M2)
receptors. The result of cholinergic transdifferentiation of sympathetic neurons
and the upregulation of M2 muscarinic receptors leads to diminished beta
adrenergic responses and could paradoxically potentiate cholinergic myocardial
responses (paradoxic bradycardia and reduced contractility) during sympathetic
nerve activation. ACh: Acetylcholine, NET: Norepinephrine transporter.
The Figure summarizes the plasticity of neurotransmitters in the sympathetic
nerve ganglion in conjunction with postsynaptic receptors on cardiac myocytes.
In the normal heart (Panel A), myocardial sympathetic nerves originating in the
stellate ganglion secrete norepinephrine (NE, red) to increase heart rate and
myocardial contractility through β1-adrenergic receptor signaling. The
interstitial NE level at the receptor is dependent upon release as well as the
presynaptic reuptake (Uptake 1) mechanism for recycling it into the presynaptic
terminal. Parasympathetic control of ventricular contractility (not illustrated)
is generally felt to play a minor role. In heart failure (Panel B), plasticity
of neurons in the sympathetic ganglia leads to transdifferentiation of some cell
bodies to a cholinergic phenotype (blue). Some sympathetic nerves now express
Choline Acetyltransferase (AChAT) and tyrosine hydroxylase (TH) expression
decreases. This transdifferentiation is promoted by Leukemia Inhibitory Factor
(LIF), Cardiotrophin-1 expression and Nerve Growth Factor (NGF) released from
the myocardium. Reductions in NE reuptake as well as increased circulating NE
levels elevate interstitial NE and lead to post-synaptic down regulation of
β1 receptor density, uncoupling of β1 receptors with G proteins
and desensitization of β adrenergic signaling. In heart failure,
cardiomyocytes paradoxically increase the expression of muscarinic (M2)
receptors. The result of cholinergic transdifferentiation of sympathetic neurons
and the upregulation of M2 muscarinic receptors leads to diminished beta
adrenergic responses and could paradoxically potentiate cholinergic myocardial
responses (paradoxic bradycardia and reduced contractility) during sympathetic
nerve activation. ACh: Acetylcholine, NET: Norepinephrine transporter.
Comment on
-
Cardiac resynchronization therapy restores sympathovagal balance in the failing heart by differential remodeling of cholinergic signaling.Circ Res. 2015 May 8;116(10):1691-9. doi: 10.1161/CIRCRESAHA.116.305268. Epub 2015 Mar 2. Circ Res. 2015. PMID: 25733594 Free PMC article.
Similar articles
-
Cardiac resynchronization therapy restores sympathovagal balance in the failing heart by differential remodeling of cholinergic signaling.Circ Res. 2015 May 8;116(10):1691-9. doi: 10.1161/CIRCRESAHA.116.305268. Epub 2015 Mar 2. Circ Res. 2015. PMID: 25733594 Free PMC article.
-
Interaction between adrenergic and cholinergic systems: presynaptic inhibitory effect of noradrenaline on acetylcholine release.J Neural Transm. 1974;Suppl 11(0):61-78. J Neural Transm. 1974. PMID: 4371488 Review. No abstract available.
-
Effect of cardiac resynchronization therapy on cardiac sympathetic nervous dysfunction and serum C-reactive protein level.Pacing Clin Electrophysiol. 2011 Oct;34(10):1225-30. doi: 10.1111/j.1540-8159.2011.03156.x. Epub 2011 Jun 14. Pacing Clin Electrophysiol. 2011. PMID: 21671958
-
Medetomidine suppresses cardiac and gastric sympathetic nerve activities but selectively activates cardiac vagus nerve.Circ J. 2014;78(6):1405-13. doi: 10.1253/circj.cj-13-1456. Epub 2014 Apr 11. Circ J. 2014. PMID: 24727611
-
Cardiac resynchronization therapy in heart failure patients with less severe left ventricular dysfunction.Eur J Heart Fail. 2015 Feb;17(2):135-43. doi: 10.1002/ejhf.208. Epub 2014 Dec 3. Eur J Heart Fail. 2015. PMID: 25469668 Review.
Cited by
-
Research progress in myocardial function and diseases related to muscarinic acetylcholine receptor (Review).Int J Mol Med. 2025 Jun;55(6):86. doi: 10.3892/ijmm.2025.5527. Epub 2025 Apr 4. Int J Mol Med. 2025. PMID: 40183403 Free PMC article. Review.
-
Guizhi Decoction () Inhibits Cholinergic Transdifferentiation by Regulating Imbalance of NGF and LIF in Salt-Sensitive Hypertensive Heart Failure Rats.Chin J Integr Med. 2020 Mar;26(3):188-196. doi: 10.1007/s11655-019-2706-6. Epub 2019 May 20. Chin J Integr Med. 2020. PMID: 31111424
-
Role of neuronal nitric oxide synthase on cardiovascular functions in physiological and pathophysiological states.Nitric Oxide. 2020 Sep 1;102:52-73. doi: 10.1016/j.niox.2020.06.004. Epub 2020 Jun 23. Nitric Oxide. 2020. PMID: 32590118 Free PMC article. Review.
-
Acute Detubulation of Ventricular Myocytes Amplifies the Inhibitory Effect of Cholinergic Agonist on Intracellular Ca2+ Transients.Front Physiol. 2021 Aug 26;12:725798. doi: 10.3389/fphys.2021.725798. eCollection 2021. Front Physiol. 2021. PMID: 34512394 Free PMC article.
-
Targeting neuro-immune systems to achieve cardiac tissue repair following myocardial infarction: A review of therapeutic approaches from in-vivo preclinical to clinical studies.Pharmacol Ther. 2023 May;245:108397. doi: 10.1016/j.pharmthera.2023.108397. Epub 2023 Mar 28. Pharmacol Ther. 2023. PMID: 36996910 Free PMC article. Review.
References
-
- Cao JM, Fishbein MC, Han JB, Lai WW, Lai AC, Wu TJ, Czer L, Wolf PL, Denton TA, Shintaku IP, Chen PS, Chen LS. Relationship between regional cardiac hyperinnervation and ventricular arrhythmia. Circulation. 2000;101:1960–1969. - PubMed
-
- Canty JM, Jr, Suzuki G, Banas MD, Verheyen F, Borgers M, Fallavollita JA. Hibernating myocardium: Chronically adapted to ischemia but vulnerable to sudden death. Circ Res. 2004;94:1142–1149. - PubMed
-
- Fallavollita JA, Heavey BM, Luisi J AJ, Michalek SM, Baldwa S, Mashtare J TL, Hutson AD, deKemp RA, Haka MS, Sajjad M, Cimato TR, Curtis AB, Cain ME, Canty J JM. Regional myocardial sympathetic denervation predicts the risk of sudden cardiac arrest in ischemic cardiomyopathy. J Am Coll Cardiol. 2014;63:141–149. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
