

Human genomics. The Genotype-Tissue Expression (GTEx) pilot analysis: multitissue gene regulation in humans
- PMID: 25954001
- PMCID: PMC4547484
- DOI: 10.1126/science.1262110
Human genomics. The Genotype-Tissue Expression (GTEx) pilot analysis: multitissue gene regulation in humans
Abstract
Understanding the functional consequences of genetic variation, and how it affects complex human disease and quantitative traits, remains a critical challenge for biomedicine. We present an analysis of RNA sequencing data from 1641 samples across 43 tissues from 175 individuals, generated as part of the pilot phase of the Genotype-Tissue Expression (GTEx) project. We describe the landscape of gene expression across tissues, catalog thousands of tissue-specific and shared regulatory expression quantitative trait loci (eQTL) variants, describe complex network relationships, and identify signals from genome-wide association studies explained by eQTLs. These findings provide a systematic understanding of the cellular and biological consequences of human genetic variation and of the heterogeneity of such effects among a diverse set of human tissues.
Copyright © 2015, American Association for the Advancement of Science.
Figures
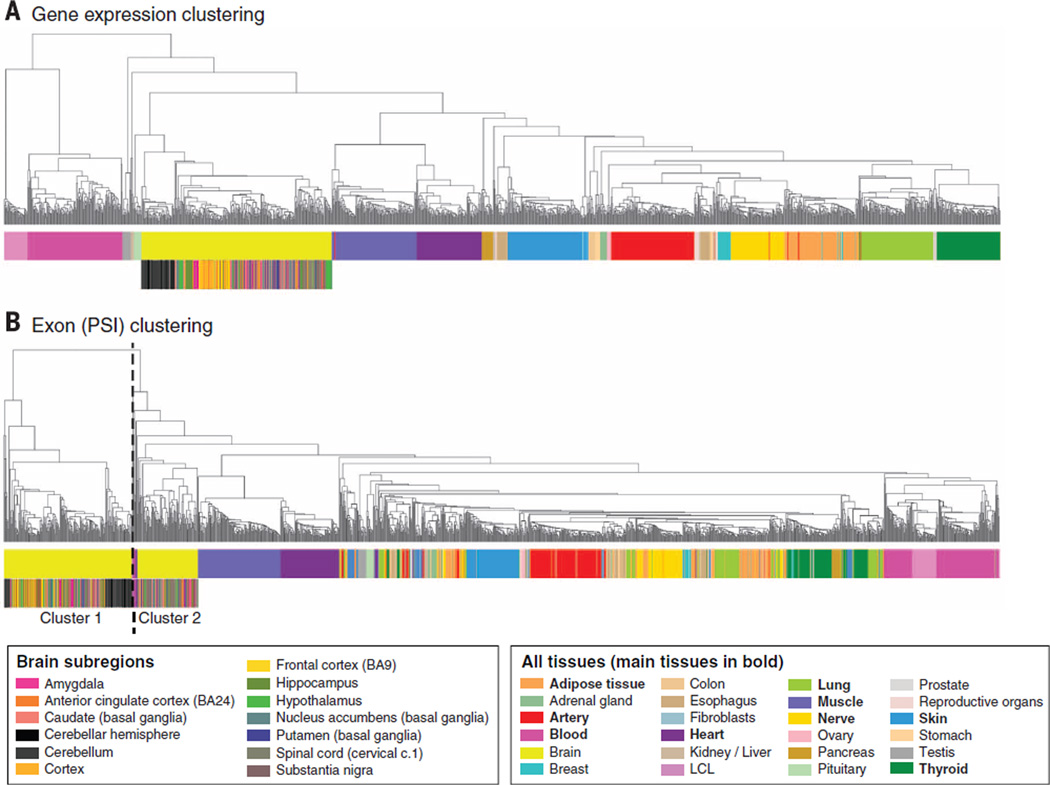
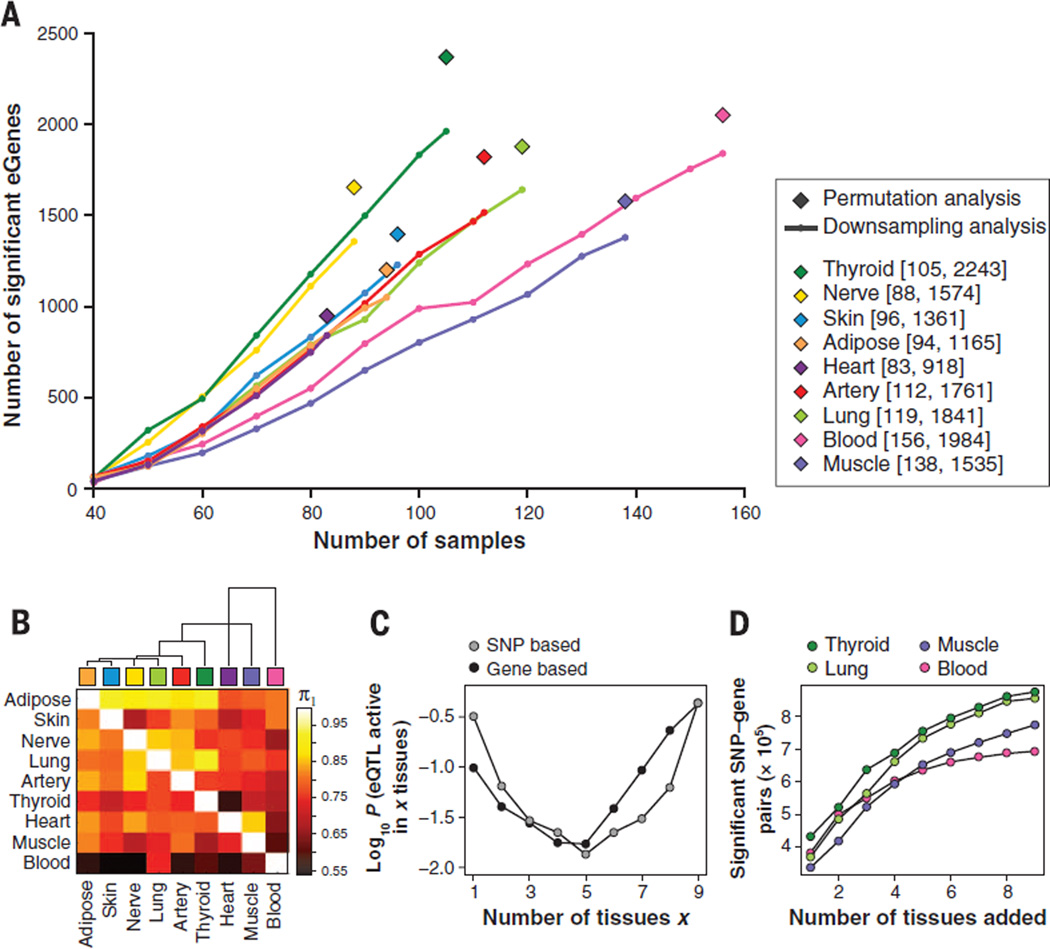
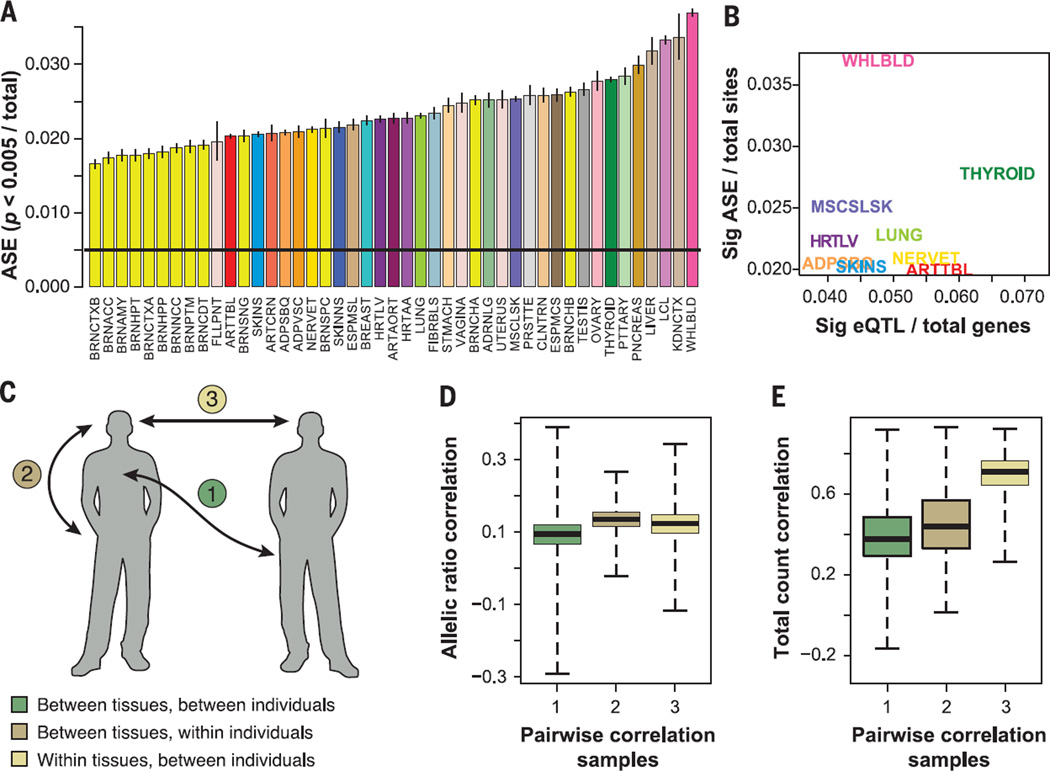
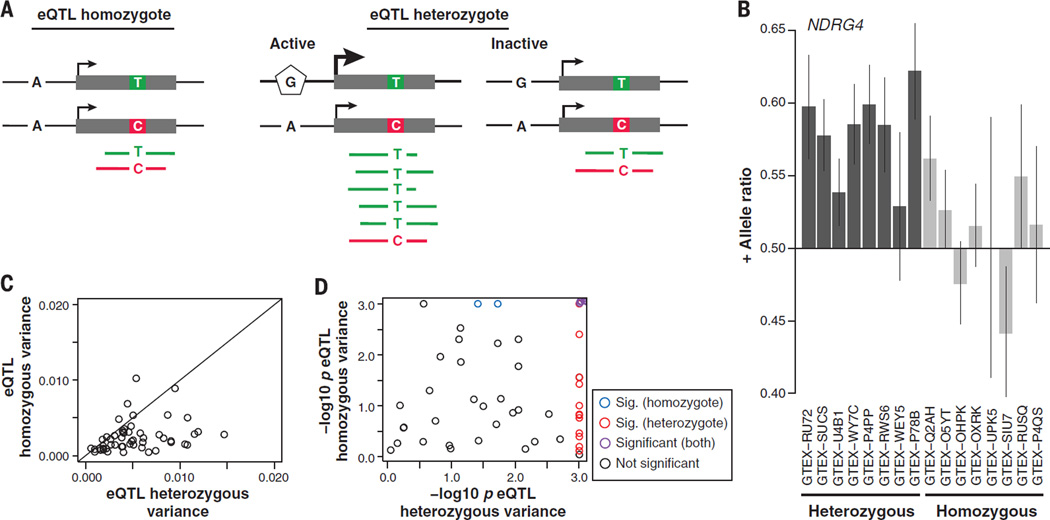
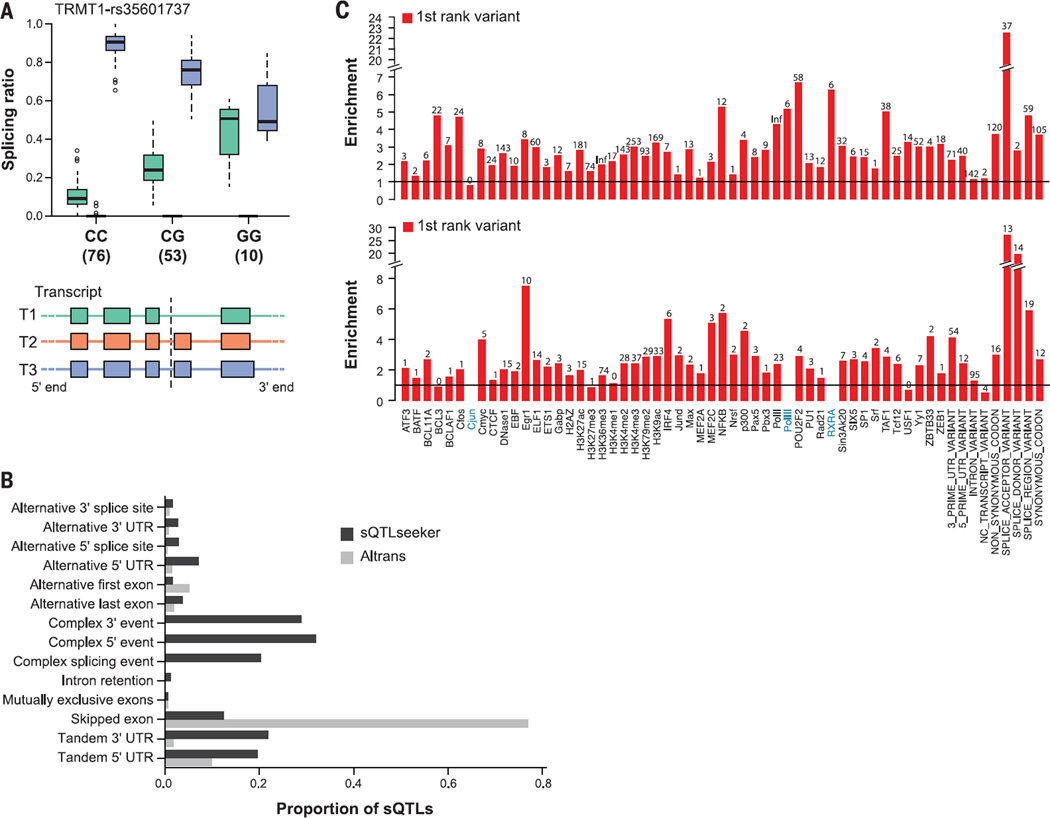
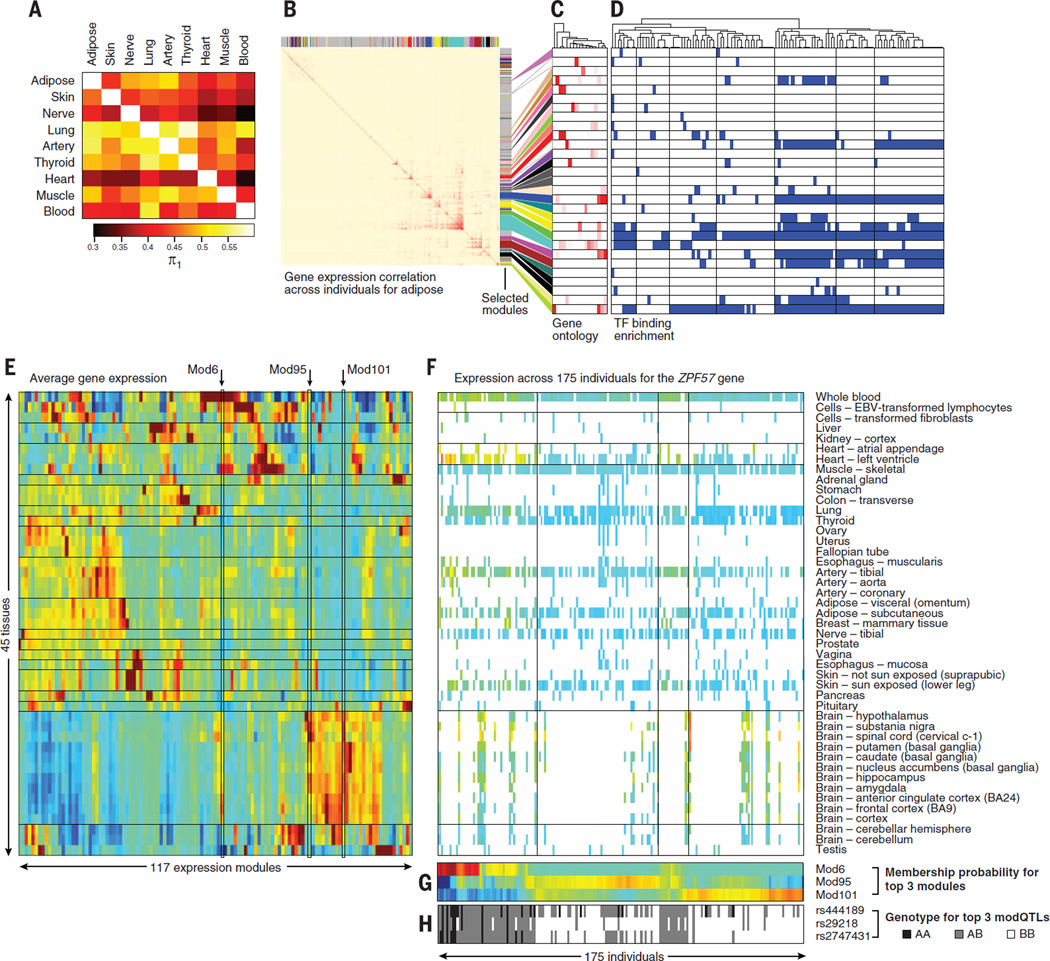
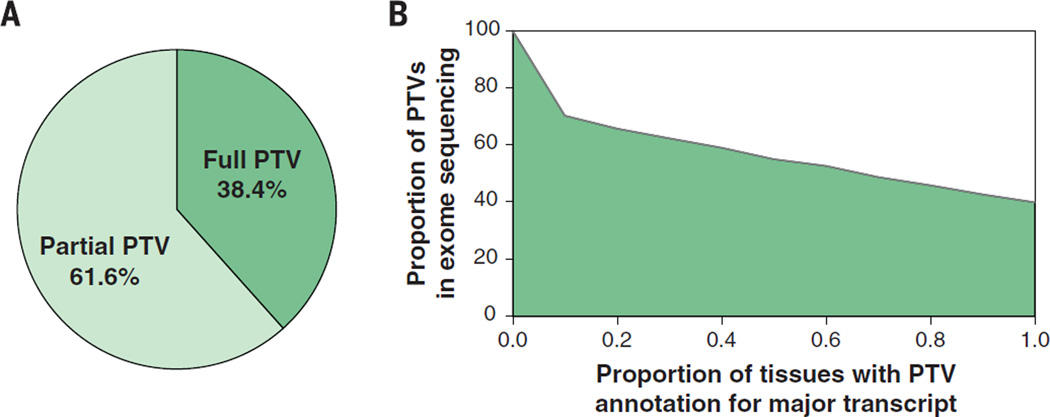
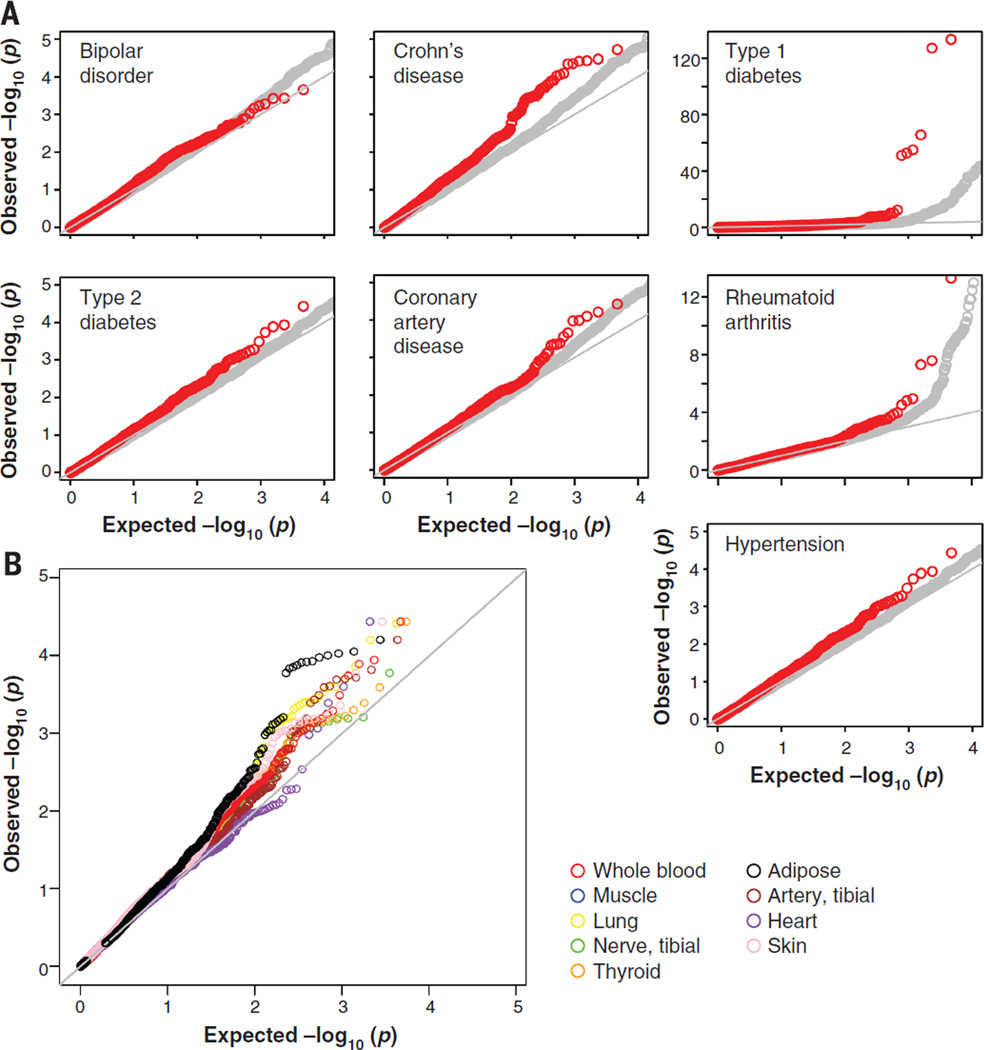
Comment in
-
Human genetics. GTEx detects genetic effects.Science. 2015 May 8;348(6235):640-1. doi: 10.1126/science.aab3002. Science. 2015. PMID: 25953996 No abstract available.
References
Publication types
MeSH terms
Substances
Associated data
Grants and funding
- 098381/WT_/Wellcome Trust/United Kingdom
- R01CA163772/CA/NCI NIH HHS/United States
- U01 HG007610/HG/NHGRI NIH HHS/United States
- U01AI111598-01/AI/NIAID NIH HHS/United States
- U01 AG046170/AG/NIA NIH HHS/United States
- R01 MH101819/MH/NIMH NIH HHS/United States
- R01 MH090936/MH/NIMH NIH HHS/United States
- U01 AI111598/AI/NIAID NIH HHS/United States
- R01 MH101820/MH/NIMH NIH HHS/United States
- WT_/Wellcome Trust/United Kingdom
- R01 CA163772/CA/NCI NIH HHS/United States
- P30 CA016056/CA/NCI NIH HHS/United States
- R01 DA006227-17/DA/NIDA NIH HHS/United States
- K12 CA139160/CA/NCI NIH HHS/United States
- R01 DA006227/DA/NIDA NIH HHS/United States
- R01AG046170/AG/NIA NIH HHS/United States
- R01 MH090951/MH/NIMH NIH HHS/United States
- R01 MH090948/MH/NIMH NIH HHS/United States
- R01 MH090941/MH/NIMH NIH HHS/United States
- HHSN261200800001C/RC/CCR NIH HHS/United States
- R01 MH090937/MH/NIMH NIH HHS/United States
- R01 GM104371/GM/NIGMS NIH HHS/United States
- HHSN268201000029C/HL/NHLBI NIH HHS/United States
- HHSN261200800001E/CA/NCI NIH HHS/United States
- P30 DK020595/DK/NIDDK NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
