

Human genomics. Effect of predicted protein-truncating genetic variants on the human transcriptome
- PMID: 25954003
- PMCID: PMC4537935
- DOI: 10.1126/science.1261877
Human genomics. Effect of predicted protein-truncating genetic variants on the human transcriptome
Abstract
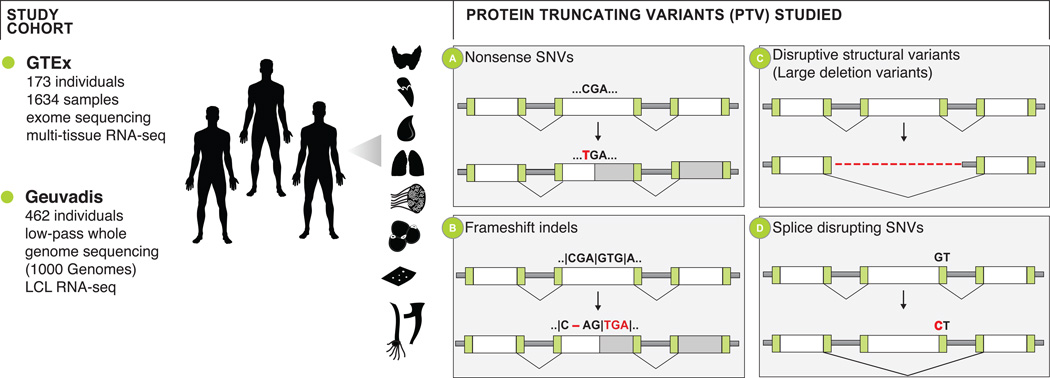
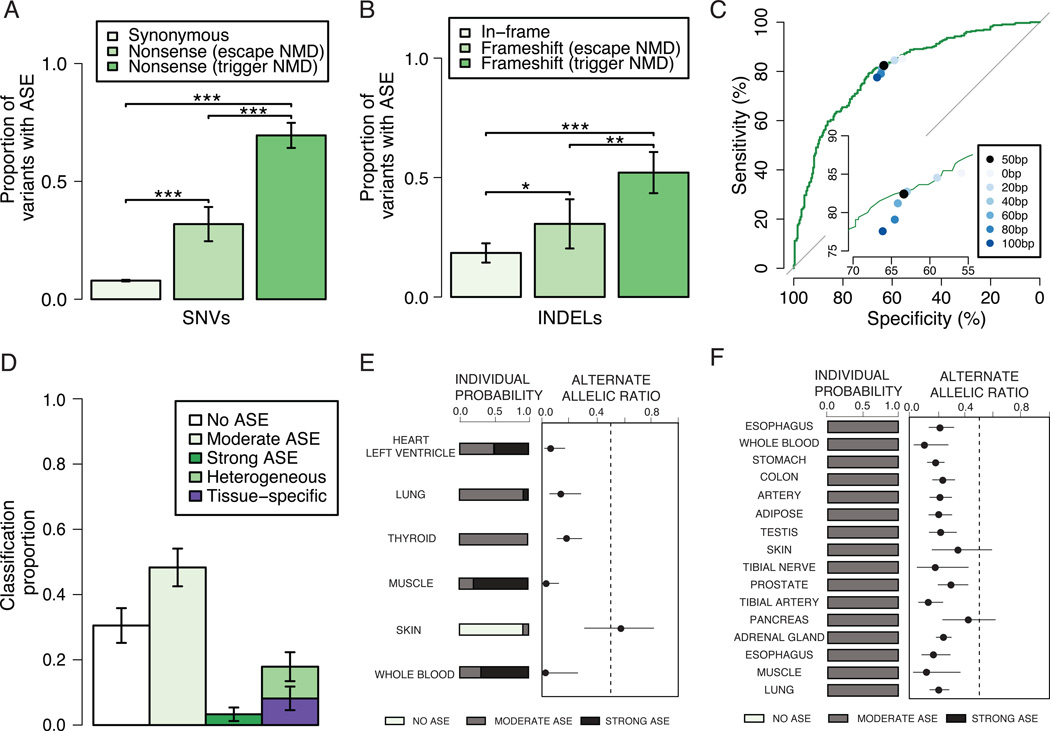
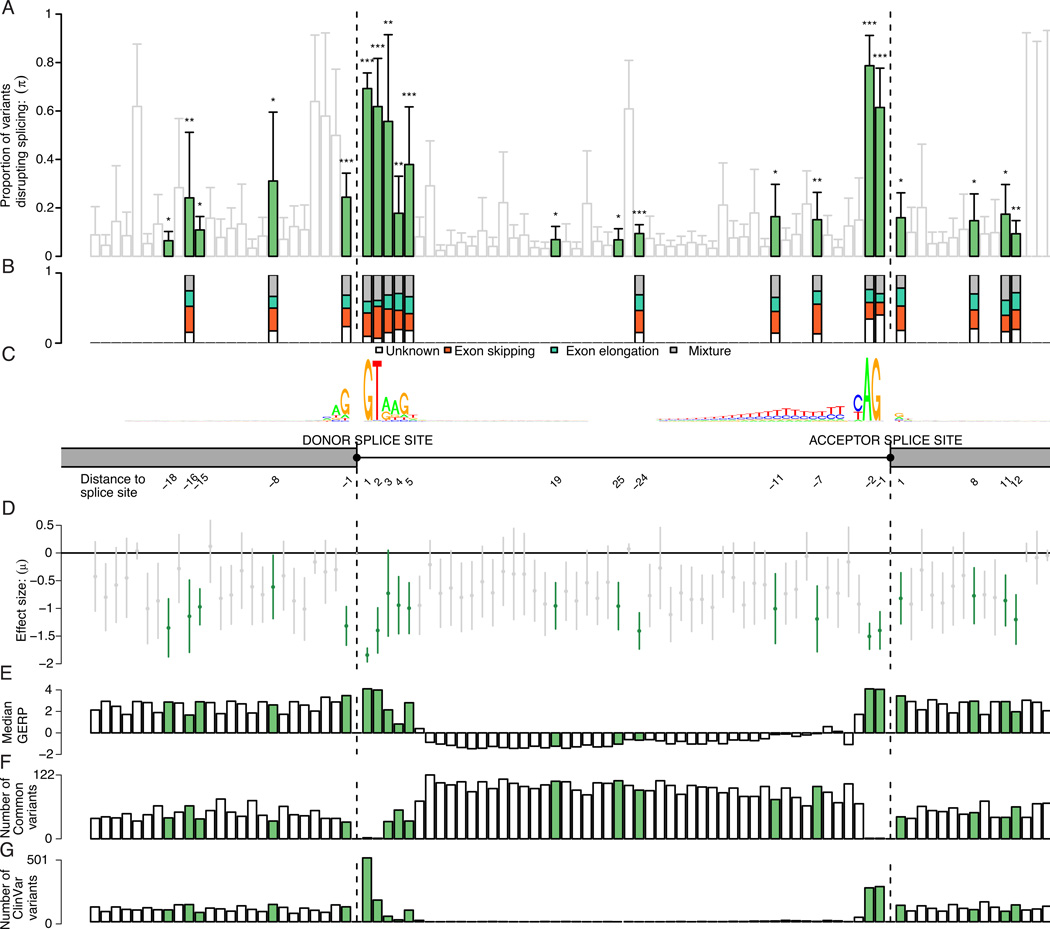
Accurate prediction of the functional effect of genetic variation is critical for clinical genome interpretation. We systematically characterized the transcriptome effects of protein-truncating variants, a class of variants expected to have profound effects on gene function, using data from the Genotype-Tissue Expression (GTEx) and Geuvadis projects. We quantitated tissue-specific and positional effects on nonsense-mediated transcript decay and present an improved predictive model for this decay. We directly measured the effect of variants both proximal and distal to splice junctions. Furthermore, we found that robustness to heterozygous gene inactivation is not due to dosage compensation. Our results illustrate the value of transcriptome data in the functional interpretation of genetic variants.
Copyright © 2015, American Association for the Advancement of Science.
Figures
Comment in
-
Human genetics. GTEx detects genetic effects.Science. 2015 May 8;348(6235):640-1. doi: 10.1126/science.aab3002. Science. 2015. PMID: 25953996 No abstract available.
References
Publication types
MeSH terms
Substances
Associated data
Grants and funding
- 098381/WT_/Wellcome Trust/United Kingdom
- R01GM104371/GM/NIGMS NIH HHS/United States
- 095552/Z/11/Z/WT_/Wellcome Trust/United Kingdom
- P30 CA016056/CA/NCI NIH HHS/United States
- U01 HG007610/HG/NHGRI NIH HHS/United States
- R01MH090941/MH/NIMH NIH HHS/United States
- R01 MH101819/MH/NIMH NIH HHS/United States
- U01 HG007593/HG/NHGRI NIH HHS/United States
- R01 MH090936/MH/NIMH NIH HHS/United States
- MH090948/MH/NIMH NIH HHS/United States
- MH090936/MH/NIMH NIH HHS/United States
- MH090941/MH/NIMH NIH HHS/United States
- T15 LM007033/LM/NLM NIH HHS/United States
- 090532/WT_/Wellcome Trust/United Kingdom
- R01 MH090951/MH/NIMH NIH HHS/United States
- MH090951/MH/NIMH NIH HHS/United States
- R01MH101814/MH/NIMH NIH HHS/United States
- 095552/WT_/Wellcome Trust/United Kingdom
- R01 DA006227/DA/NIDA NIH HHS/United States
- MH090937/MH/NIMH NIH HHS/United States
- R01 MH101810/MH/NIMH NIH HHS/United States
- DA006227/DA/NIDA NIH HHS/United States
- 090532/Z/09/Z/WT_/Wellcome Trust/United Kingdom
- R01 MH101820/MH/NIMH NIH HHS/United States
- R01MH101810/MH/NIMH NIH HHS/United States
- R01 MH090948/MH/NIMH NIH HHS/United States
- R01 MH090941/MH/NIMH NIH HHS/United States
- HHSN261200800001C/RC/CCR NIH HHS/United States
- R01 MH090937/MH/NIMH NIH HHS/United States
- R01 GM104371/GM/NIGMS NIH HHS/United States
- HHSN268201000029C/HL/NHLBI NIH HHS/United States
- U01HG007593/HG/NHGRI NIH HHS/United States
- HHSN261200800001E/CA/NCI NIH HHS/United States
- R01 MH101814/MH/NIMH NIH HHS/United States
- P30 DK020595/DK/NIDDK NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
