

Characterization of early disease status in treatment-naive male paediatric patients with Fabry disease enrolled in a randomized clinical trial
- PMID: 25955246
- PMCID: PMC4425695
- DOI: 10.1371/journal.pone.0124987
Characterization of early disease status in treatment-naive male paediatric patients with Fabry disease enrolled in a randomized clinical trial
Abstract
Trial design: This analysis characterizes the degree of early organ involvement in a cohort of oligo-symptomatic untreated young patients with Fabry disease enrolled in an ongoing randomized, open-label, parallel-group, phase 3B clinical trial.
Methods: Males aged 5-18 years with complete α-galactosidase A deficiency, without symptoms of major organ damage, were enrolled in a phase 3B trial evaluating two doses of agalsidase beta. Baseline disease characteristics of 31 eligible patients (median age 12 years) were studied, including cellular globotriaosylceramide (GL-3) accumulation in skin (n = 31) and kidney biopsy (n = 6; median age 15 years; range 13-17 years), renal function, and glycolipid levels (plasma, urine).
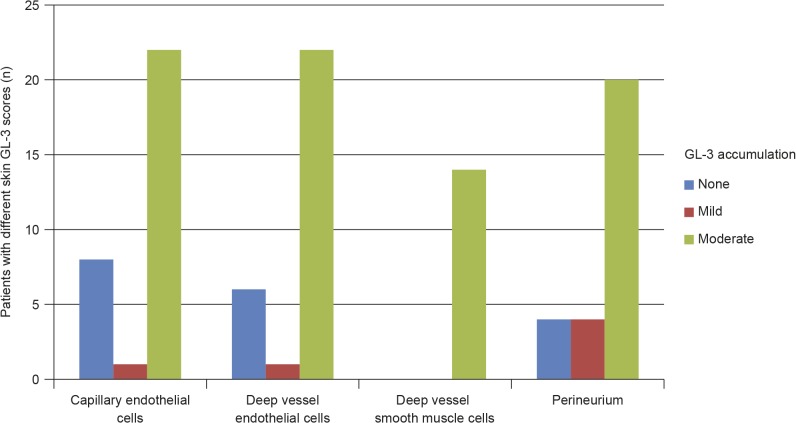
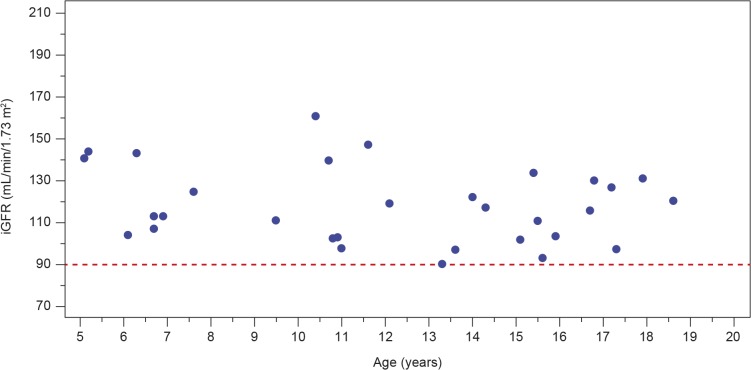
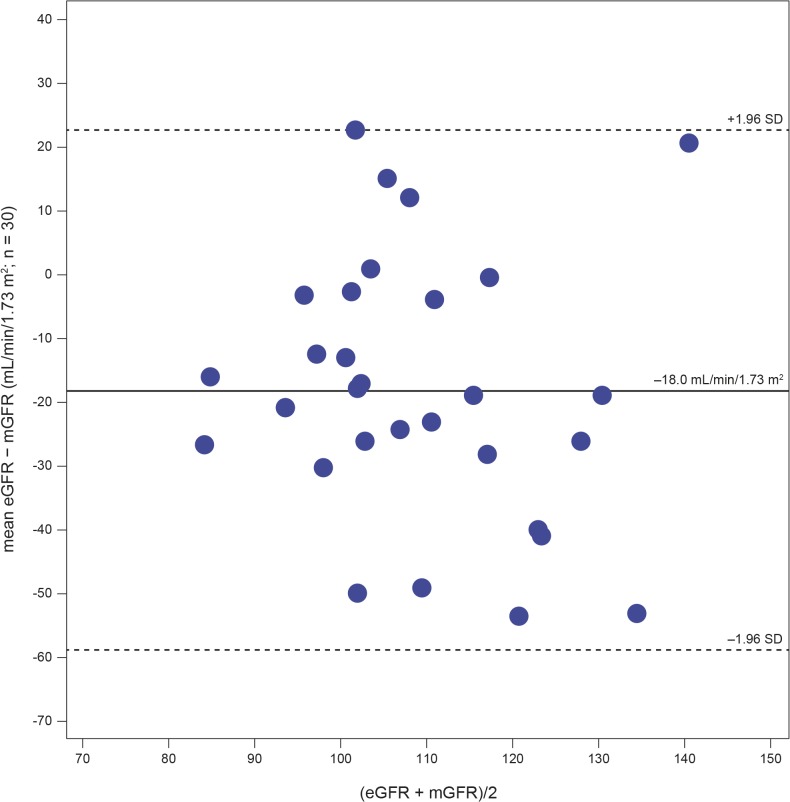
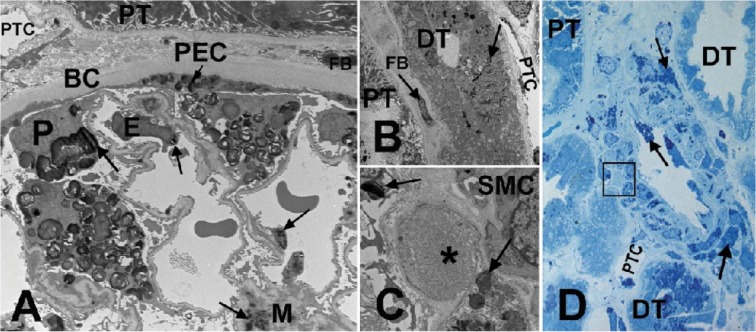
Results: Plasma and urinary GL-3 levels were abnormal in 25 of 30 and 31 of 31 patients, respectively. Plasma lyso-GL-3 was elevated in all patients. GL-3 accumulation was documented in superficial skin capillary endothelial cells (23/31 patients) and deep vessel endothelial cells (23/29 patients). The mean glomerular filtration rate (GFR), measured by plasma disappearance of iohexol, was 118.1 mL/min/1.73 m(2) (range 90.4-161.0 mL/min/1.73 m(2)) and the median urinary albumin/creatinine ratio was 10 mg/g (range 4.0-27.0 mg/g). On electron microscopy, renal biopsy revealed GL-3 accumulation in all glomerular cell types (podocytes and parietal, endothelial, and mesangial cells), as well as in peritubular capillary and non-capillary endothelial, interstitial, vascular smooth muscle, and distal tubules/collecting duct cells. Lesions indicative of early Fabry arteriopathy and segmental effacement of podocyte foot processes were found in all 6 patients.
Conclusions: These data reveal that in this small cohort of children with Fabry disease, histological evidence of GL-3 accumulation, and cellular and vascular injury are present in renal tissues at very early stages of the disease, and are noted before onset of microalbuminuria and development of clinically significant renal events (e.g. reduced GFR). These data give additional support to the consideration of early initiation of enzyme replacement therapy, potentially improving long-term outcome.
Trial registration: ClinicalTrials.gov NCT00701415.
Conflict of interest statement
Figures
References
-
- Desnick RJ, Ioannou YA, Eng CM. Alpha-galactosidase A deficiency: Fabry Disease In: Scriver C, Beaudet A, Sly W, Valle D, editors. The metabolic bases of inherited disease. 8th ed. New York: McGraw-Hill; 2001. pp. 3733–3774.
-
- Breunig F, Wanner C. Enzyme replacement therapy for Fabry disease: proving the clinical benefit. Nephrol Dial Transplant. 2003;18: 7–9. - PubMed
-
- Eng CM, Germain DP, Banikazemi M, Warnock DG, Wanner C, Hopkin RJ, et al. Fabry disease: guidelines for the evaluation and management of multi-organ system involvement. Genet Med. 2006;8: 539–548. - PubMed
-
- Wilcox WR, Oliviera JP, Hopkin RJ, Ortiz A, Banikazemi M, Feldt-Rasmussen U, et al. Females with Fabry disease frequently have major organ involvement: lessons from the Fabry registry. Mol Genet Metab. 2008;93: 112–128. - PubMed
Publication types
MeSH terms
Substances
Associated data
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
