

Multiple Mechanisms of Unfolded Protein Response-Induced Cell Death
- PMID: 25956028
- PMCID: PMC4484218
- DOI: 10.1016/j.ajpath.2015.03.009
Multiple Mechanisms of Unfolded Protein Response-Induced Cell Death
Abstract
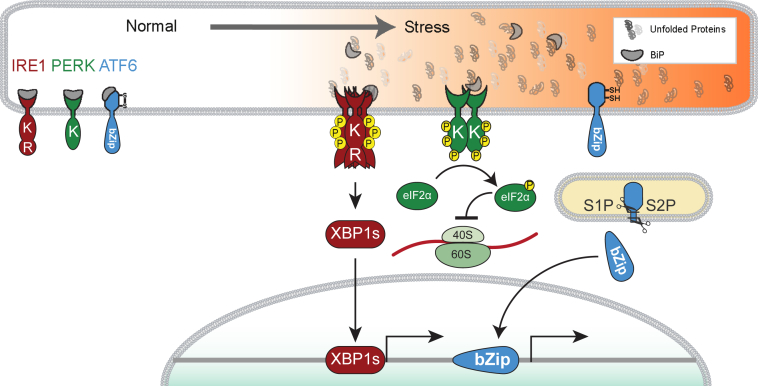
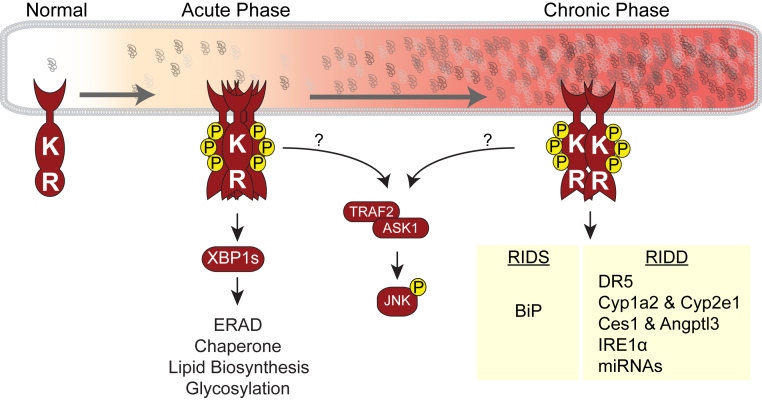
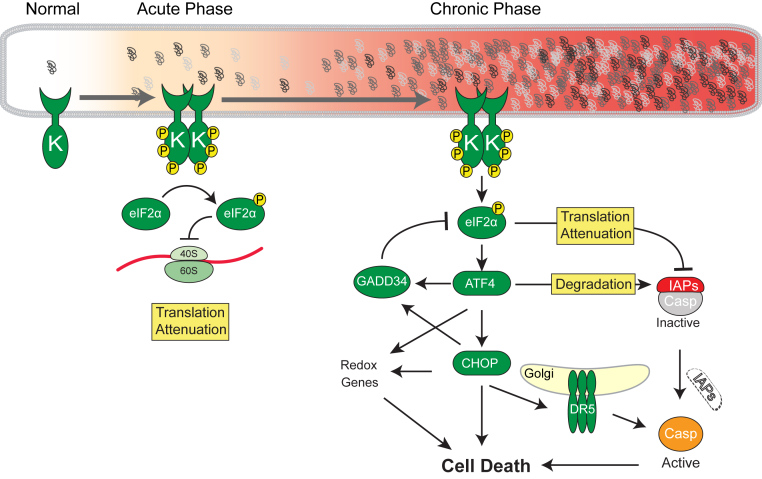
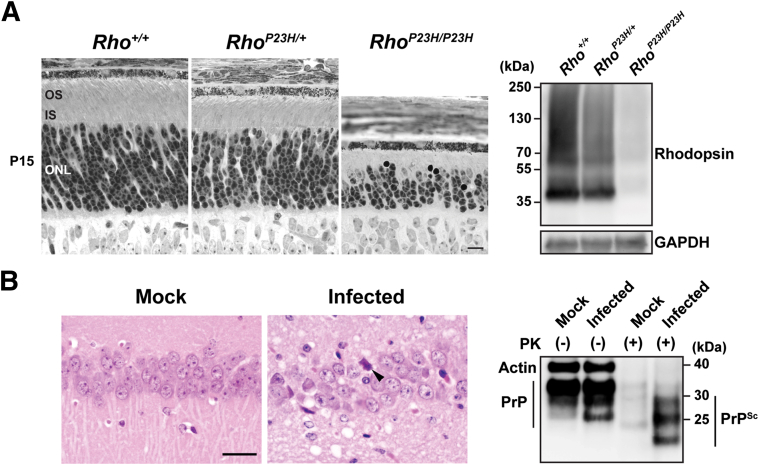
Eukaryotic cells fold and assemble membrane and secreted proteins in the endoplasmic reticulum (ER), before delivery to other cellular compartments or the extracellular environment. Correctly folded proteins are released from the ER, and poorly folded proteins are retained until they achieve stable conformations; irreparably misfolded proteins are targeted for degradation. Diverse pathological insults, such as amino acid mutations, hypoxia, or infection, can overwhelm ER protein quality control, leading to misfolded protein buildup, causing ER stress. To cope with ER stress, eukaryotic cells activate the unfolded protein response (UPR) by increasing levels of ER protein-folding enzymes and chaperones, enhancing the degradation of misfolded proteins, and reducing protein translation. In mammalian cells, three ER transmembrane proteins, inositol-requiring enzyme-1 (IRE1; official name ERN1), PKR-like ER kinase (PERK; official name EIF2AK3), and activating transcription factor-6, control the UPR. The UPR signaling triggers a set of prodeath programs when the cells fail to successfully adapt to ER stress or restore homeostasis. ER stress and UPR signaling are implicated in the pathogenesis of diverse diseases, including neurodegeneration, cancer, diabetes, and inflammation. This review discusses the current understanding in both adaptive and apoptotic responses as well as the molecular mechanisms instigating apoptosis via IRE1 and PERK signaling. We also examine how IRE1 and PERK signaling may be differentially used during neurodegeneration arising in retinitis pigmentosa and prion infection.
Copyright © 2015 American Society for Investigative Pathology. Published by Elsevier Inc. All rights reserved.
Figures
References
-
- Cox J.S., Shamu C.E., Walter P. Transcriptional induction of genes encoding endoplasmic reticulum resident proteins requires a transmembrane protein kinase. Cell. 1993;73:1197–1206. - PubMed
-
- Calfon M., Zeng H., Urano F., Till J.H., Hubbard S.R., Harding H.P., Clark S.G., Ron D. IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature. 2002;415:92–96. - PubMed
-
- Yoshida H., Matsui T., Yamamoto A., Okada T., Mori K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell. 2001;107:881–891. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
