

Comparative transcriptome profiling approach to glean virulence and immunomodulation-related genes of Fasciola hepatica
- PMID: 25956885
- PMCID: PMC4429430
- DOI: 10.1186/s12864-015-1539-8
Comparative transcriptome profiling approach to glean virulence and immunomodulation-related genes of Fasciola hepatica
Abstract
Background: Fasciola hepatica causes chronic liver disease, fasciolosis, leading to significant losses in the livestock economy and concerns for human health in many countries. The identification of F. hepatica genes involved in the parasite's virulence through modulation of host immune system is utmost important to comprehend evasion mechanisms of the parasite and develop more effective strategies against fasciolosis. In this study, to identify the parasite's putative virulence genes which are associated with host immunomodulation, we explored whole transcriptome of an adult F. hepatica using current transcriptome profiling approaches integrated with detailed in silico analyses. In brief, the comparison of the parasite transcripts with the specialised public databases containing sequence data of non-parasitic organisms (Dugesiidae species and Caenorhabditis elegans) or of numerous pathogens and investigation of the sequences in terms of nucleotide evolution (directional selection) and cytokine signaling relation were conducted.
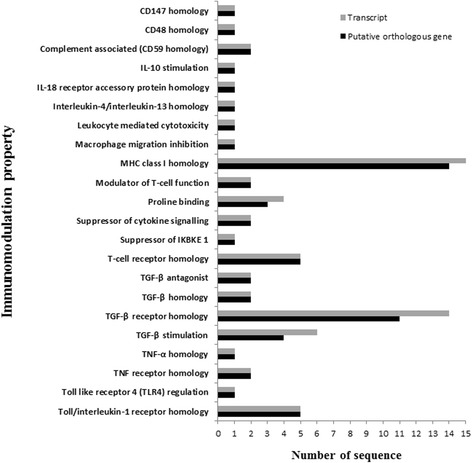
Results: NGS of the whole transcriptome resulted in 19,534,766 sequence reads, yielding a total of 40,260 transcripts (N₅₀ = 522 bp). A number of the parasite transcripts (n = 1,671) were predicted to be virulence-related on the basis of the exclusive homology with the pathogen-associated data, positive selection or relationship with cytokine signaling. Of these, a group of the virulence-related genes (n = 62), not previously described, were found likely to be associated with immunomodulation based on in silico functional categorisation, showing significant sequence similarities with various immune receptors (i.e. MHC I class, TGF-β receptor, toll/interleukin-1 receptor, T-cell receptor, TNF receptor, and IL-18 receptor accessory protein), cytokines (i.e. TGF-β, interleukin-4/interleukin-13 and TNF-α), cluster of differentiations (e.g. CD48 and CD147) or molecules associated with other immunomodulatory mechanisms (such as regulation of macrophage activation). Some of the genes (n = 5) appeared to be under positive selection (Ka/Ks > 1), imitating proteins associated with cytokine signaling (through sequence homologies with thrombospondin type 1, toll/interleukin-1 receptor, TGF-β receptor and CD147).
Conclusions: With a comparative transcriptome profiling approach, we have identified a number of potential immunomodulator genes of F. hepatica (n = 62), which are firstly described here, could be employed for the development of better strategies (including RNAi) in the battle against both zoonotically and economically important disease, fasciolosis.
Figures
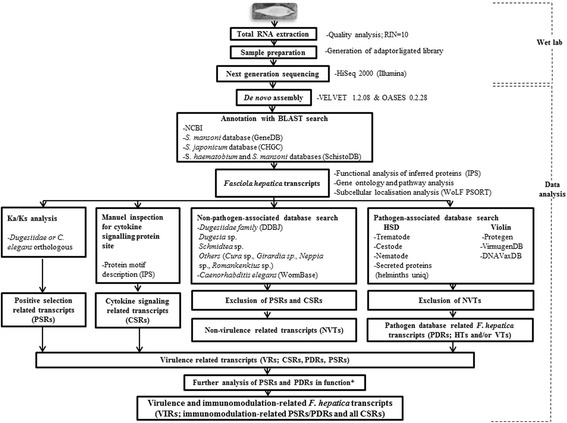
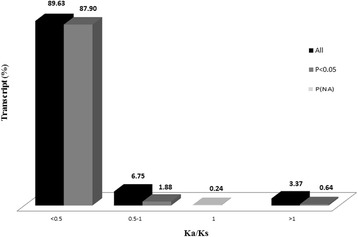
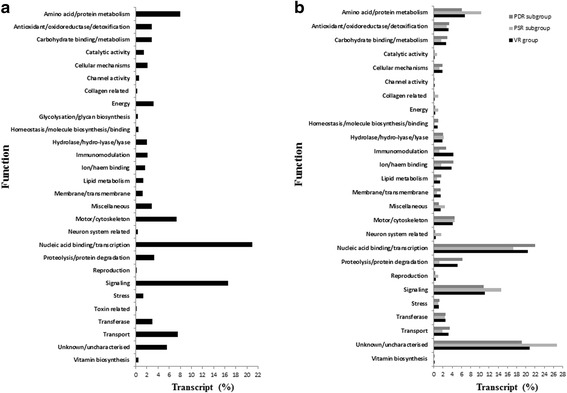
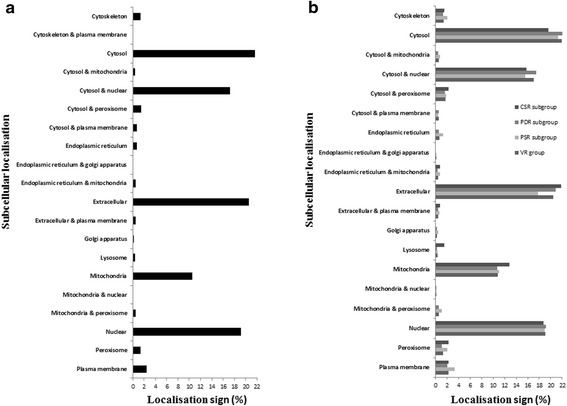
References
-
- Mas-Coma S, Agramunt VH, Valero MA. Neurological and ocular fascioliasis in humans. Adv Parasitol. 2014;84:27–149. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
