

Autosomal-Dominant Multiple Pterygium Syndrome Is Caused by Mutations in MYH3
- PMID: 25957469
- PMCID: PMC4570285
- DOI: 10.1016/j.ajhg.2015.04.004
Autosomal-Dominant Multiple Pterygium Syndrome Is Caused by Mutations in MYH3
Abstract
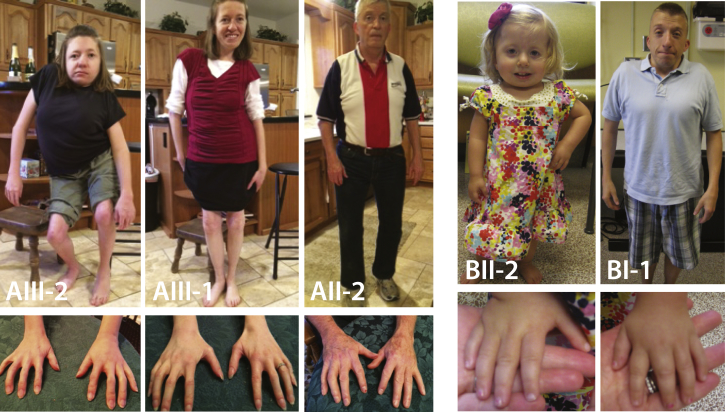
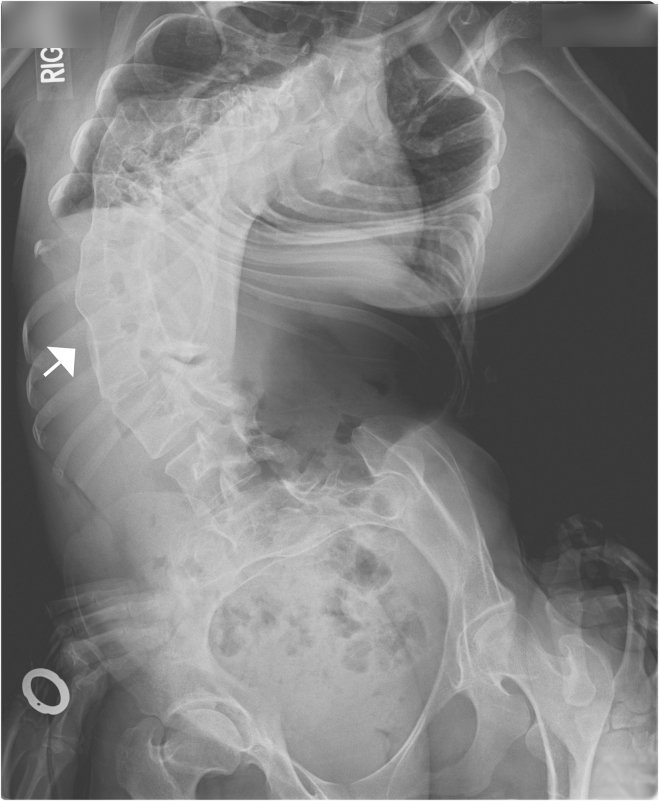
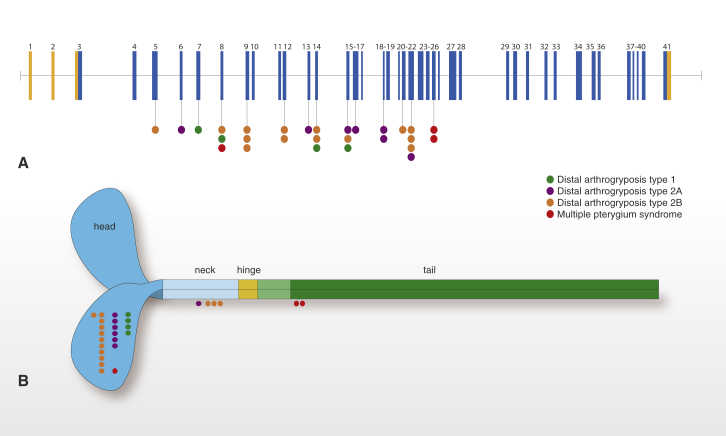
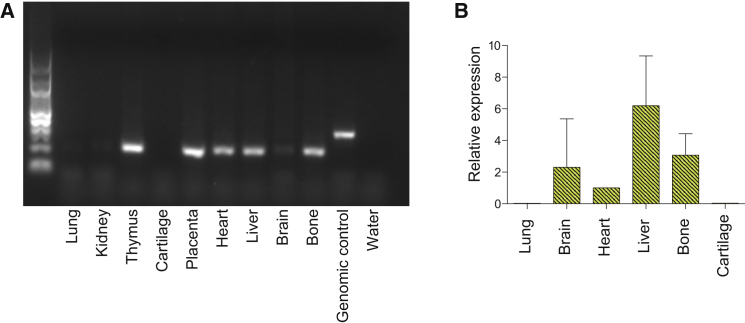
Multiple pterygium syndrome (MPS) is a phenotypically and genetically heterogeneous group of rare Mendelian conditions characterized by multiple pterygia, scoliosis, and congenital contractures of the limbs. MPS typically segregates as an autosomal-recessive disorder, but rare instances of autosomal-dominant transmission have been reported. Whereas several mutations causing recessive MPS have been identified, the genetic basis of dominant MPS remains unknown. We identified four families affected by dominantly transmitted MPS characterized by pterygia, camptodactyly of the hands, vertebral fusions, and scoliosis. Exome sequencing identified predicted protein-altering mutations in embryonic myosin heavy chain (MYH3) in three families. MYH3 mutations underlie distal arthrogryposis types 1, 2A, and 2B, but all mutations reported to date occur in the head and neck domains. In contrast, two of the mutations found to cause MPS in this study occurred in the tail domain. The phenotypic overlap among persons with MPS, coupled with physical findings distinct from other conditions caused by mutations in MYH3, suggests that the developmental mechanism underlying MPS differs from that of other conditions and/or that certain functions of embryonic myosin might be perturbed by disruption of specific residues and/or domains. Moreover, the vertebral fusions in persons with MPS, coupled with evidence of MYH3 expression in bone, suggest that embryonic myosin plays a role in skeletal development.
Copyright © 2015 The American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures
References
-
- Vogt J., Morgan N.V., Rehal P., Faivre L., Brueton L.A., Becker K., Fryns J.P., Holder S., Islam L., Kivuva E. CHRNG genotype-phenotype correlations in the multiple pterygium syndromes. J. Med. Genet. 2012;49:21–26. - PubMed
-
- Frias, J.L., Holahan, J.R., Rosenbloom, A.L., and Felman, A.H. (1973). An autosomal dominant syndrome of multiple pterygium, ptosis, and skeletal abnormalities. Proceedings of the Fourth International Conference on Birth Defects. Excerpta Medica 19.
-
- Kawira E.L., Bender H.A. An unusual distal arthrogryposis. Am. J. Med. Genet. 1985;20:425–429. - PubMed
-
- Prontera P., Sensi A., Merlo L., Garani G., Cocchi G., Calzolari E. Familial occurrence of multiple pterygium syndrome: expression in a heterozygote of the recessive form or variability of the dominant form? Am. J. Med. Genet. A. 2006;140:2227–2230. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
- U54 HG006493/HG/NHGRI NIH HHS/United States
- HD024064/HD/NICHD NIH HHS/United States
- P30 HD024064/HD/NICHD NIH HHS/United States
- P30 DK056338/DK/NIDDK NIH HHS/United States
- R01 HD048895/HD/NICHD NIH HHS/United States
- UM1 HG006493/HG/NHGRI NIH HHS/United States
- R01 AR066124/AR/NIAMS NIH HHS/United States
- RC2 HG005608/HG/NHGRI NIH HHS/United States
- T32GM07526/GM/NIGMS NIH HHS/United States
- 1R01HD048895/HD/NICHD NIH HHS/United States
- 1RC2HG005608/HG/NHGRI NIH HHS/United States
- U54HG003273/HG/NHGRI NIH HHS/United States
- R00 HG004316/HG/NHGRI NIH HHS/United States
- U54 HG006542/HG/NHGRI NIH HHS/United States
- U54HD061221/HD/NICHD NIH HHS/United States
- T32 GM007526/GM/NIGMS NIH HHS/United States
- U54 HD083092/HD/NICHD NIH HHS/United States
- U54 HD061221/HD/NICHD NIH HHS/United States
- U54 U54HG006542/HG/NHGRI NIH HHS/United States
- P01HD070394/HD/NICHD NIH HHS/United States
- U54 HG003273/HG/NHGRI NIH HHS/United States
- 5R000HG004316/HG/NHGRI NIH HHS/United States
- R01AR066124/AR/NIAMS NIH HHS/United States
- 1U54HG006493/HG/NHGRI NIH HHS/United States
- P01 HD070394/HD/NICHD NIH HHS/United States
- UL1 TR001067/TR/NCATS NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
