

Long-term outcomes in Ornithine Transcarbamylase deficiency: a series of 90 patients
- PMID: 25958381
- PMCID: PMC4443534
- DOI: 10.1186/s13023-015-0266-1
Long-term outcomes in Ornithine Transcarbamylase deficiency: a series of 90 patients
Abstract
Background: The principal aim of this study was to investigate the long-term outcomes of a large cohort of patients with ornithine transcarbamylase deficiency (OTCD) who were followed up at a single medical center.
Methods: We analyzed clinical, biochemical and genetic parameters of 90 patients (84 families, 48 males and 42 females) with OTCD between 1971 and 2011.
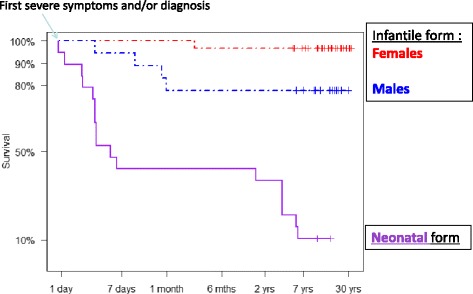
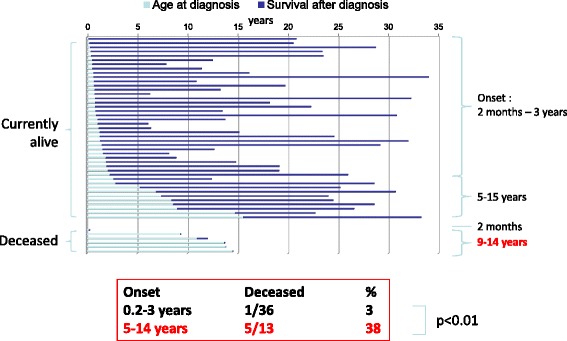
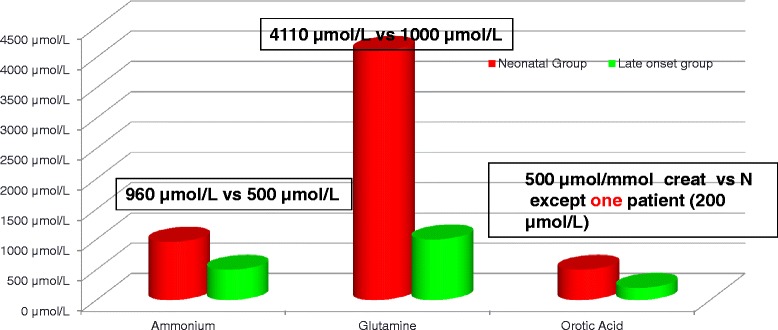
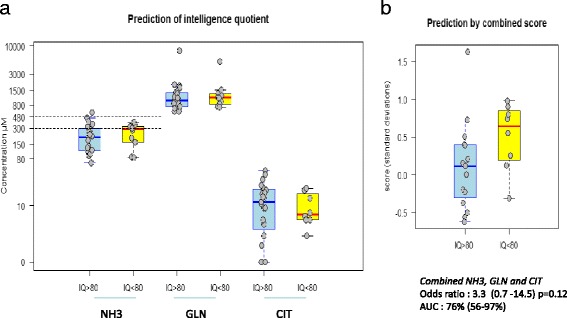
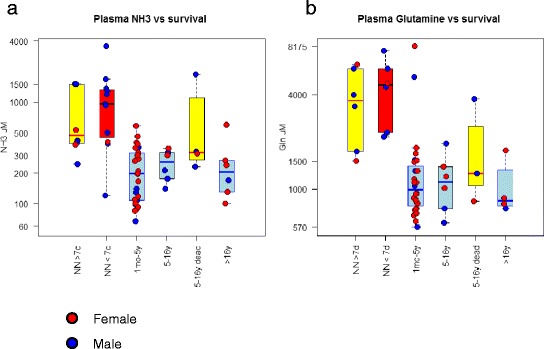
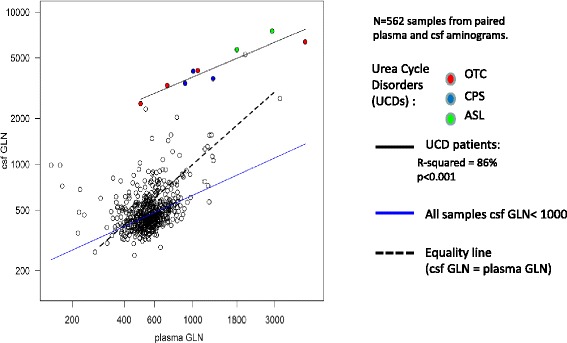
Results: Twenty-seven patients (22 boys, 5 girls) had a neonatal presentation; 52 patients had an "intermediate" late-onset form of the disease (21 boys, 31 girls) that was revealed between 1 month and 16 years; and 11 patients (5 boys, 6 girls) presented in adulthood (16 to 55 years). Patients with a neonatal presentation had increased mortality (90% versus 13% in late-onset forms) and peak plasma ammonium (mean value: 960 μmol/L versus 500 μmol/L) and glutamine (mean value: 4110 μmol/L versus 1000 μmol/L) levels at diagnosis. All of the neonatal forms displayed a greater number of acute decompensations (mean value: 6.2/patient versus 2.5 and 1.4 in infants and adults, respectively). In the adult group, some patients even recently died at the time of presentation during their first episode of coma. Molecular analyses identified a deleterious mutation in 59/68 patients investigated. Single base substitutions were detected more frequently than deletions (69% and 12%, respectively), with a recurrent mutation identified in the late-onset groups (pArg40 His; 13% in infants, 57% in adults); inherited mutations represented half of the cases. The neurological score did not differ significantly between the patients who were alive in the neonatal or late-onset groups and did not correlate with the peak ammonia and plasma glutamine concentrations at diagnosis. However, in late-onset forms of the disease, ammonia levels adjusted according to the glutamine/citrulline ratio at diagnosis were borderline predictors of low IQ (p = 0.12 by logistic regression; area under the receiver operating characteristic curve of 76%, p <0.05).
Conclusions: OTCD remains a severe disease, even in adult-onset patients for whom the prevention of metabolic decompensations is crucial. The combination of biochemical markers warrants further investigations to provide additional prognostic information regarding the neurological outcomes of patients with OTCD.
Figures
References
-
- Brusilow SW, Maestri NE. Urea cycle disorders: diagnosis, pathophysiology, and therapy. Adv Pediatr. 1996;43:127–70. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
