

Principles of long noncoding RNA evolution derived from direct comparison of transcriptomes in 17 species
- PMID: 25959816
- PMCID: PMC4576741
- DOI: 10.1016/j.celrep.2015.04.023
Principles of long noncoding RNA evolution derived from direct comparison of transcriptomes in 17 species
Abstract
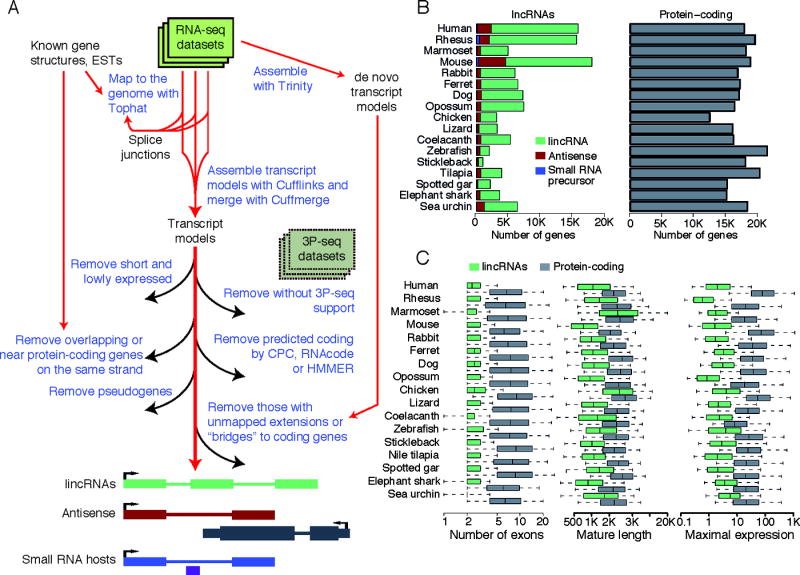
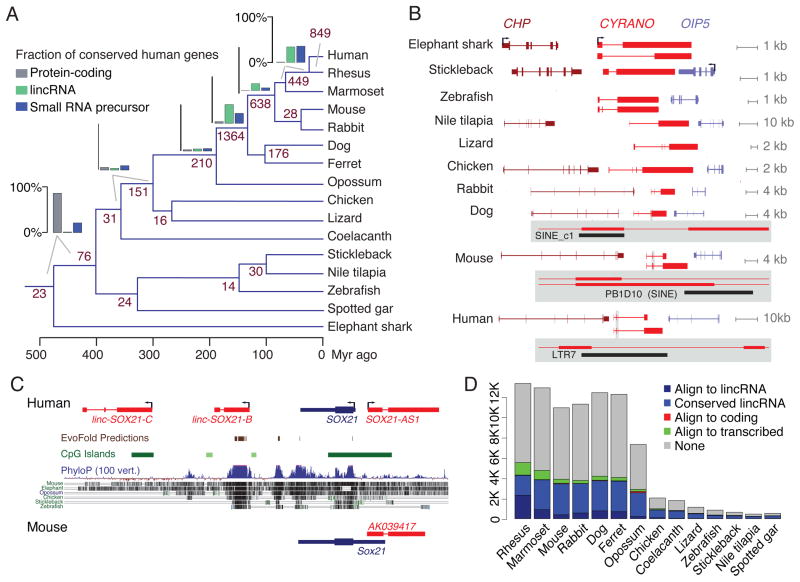
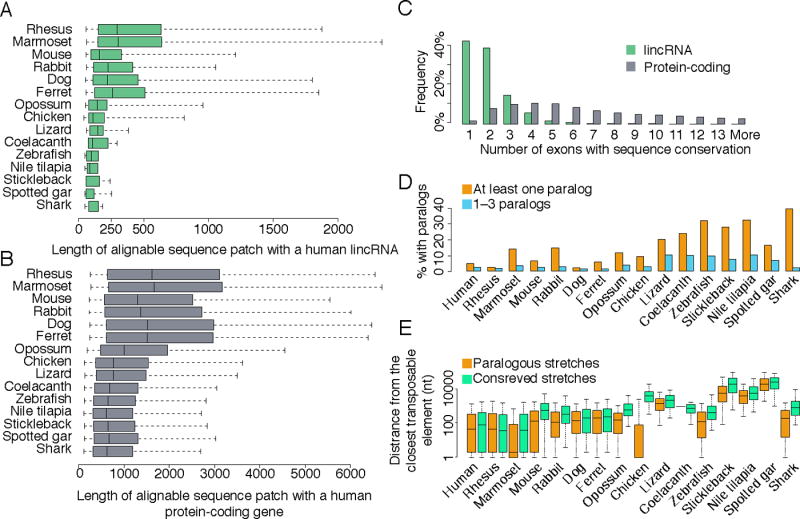
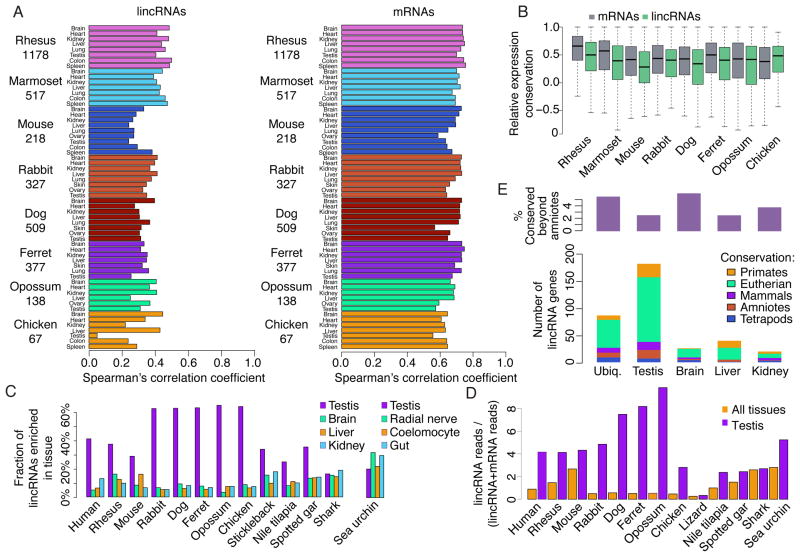
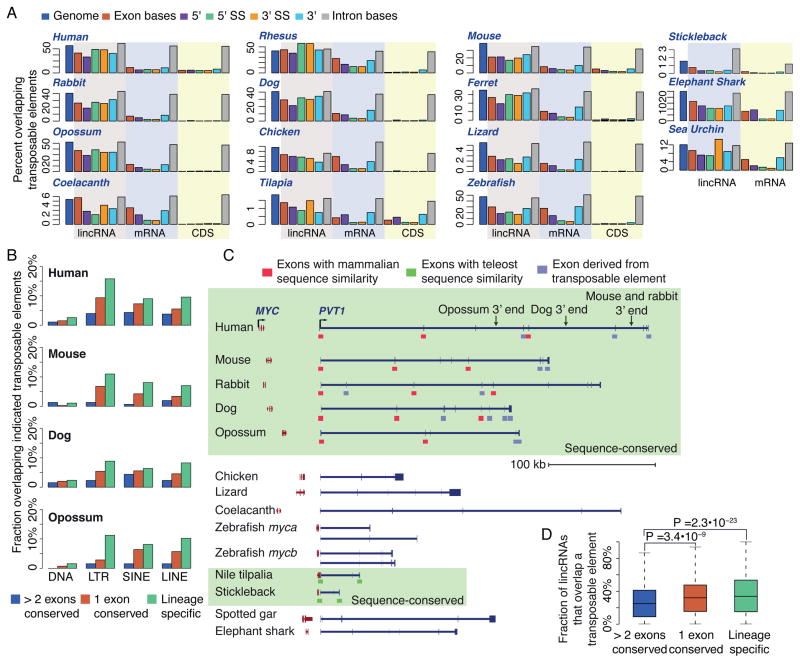
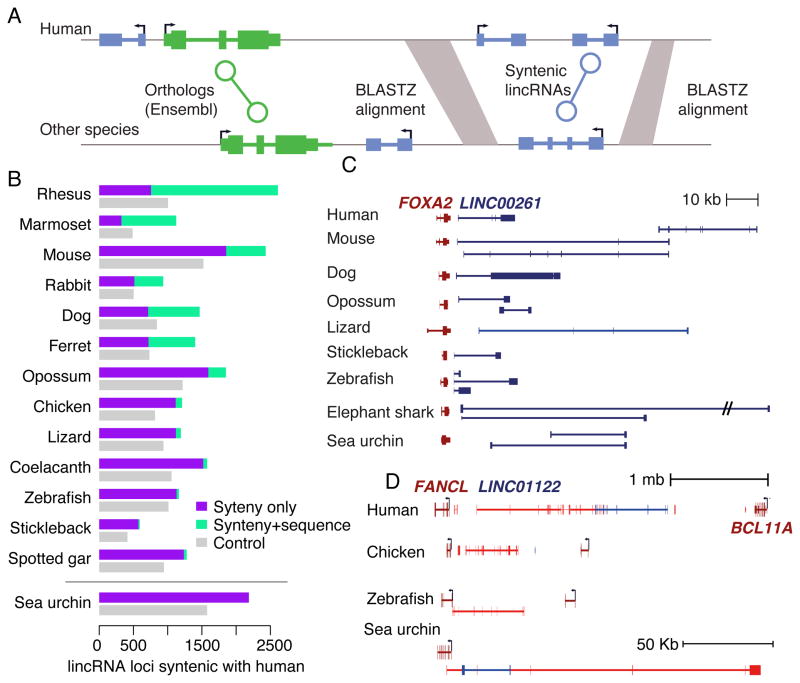
The inability to predict long noncoding RNAs from genomic sequence has impeded the use of comparative genomics for studying their biology. Here, we develop methods that use RNA sequencing (RNA-seq) data to annotate the transcriptomes of 16 vertebrates and the echinoid sea urchin, uncovering thousands of previously unannotated genes, most of which produce long intervening noncoding RNAs (lincRNAs). Although in each species, >70% of lincRNAs cannot be traced to homologs in species that diverged >50 million years ago, thousands of human lincRNAs have homologs with similar expression patterns in other species. These homologs share short, 5'-biased patches of sequence conservation nested in exonic architectures that have been extensively rewired, in part by transposable element exonization. Thus, over a thousand human lincRNAs are likely to have conserved functions in mammals, and hundreds beyond mammals, but those functions require only short patches of specific sequences and can tolerate major changes in gene architecture.
Copyright © 2015 The Authors. Published by Elsevier Inc. All rights reserved.
Figures
References
Publication types
MeSH terms
Substances
Associated data
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
