

Pan-cancer stratification of solid human epithelial tumors and cancer cell lines reveals commonalities and tissue-specific features of the CpG island methylator phenotype
- PMID: 25960768
- PMCID: PMC4424513
- DOI: 10.1186/s13072-015-0007-7
Pan-cancer stratification of solid human epithelial tumors and cancer cell lines reveals commonalities and tissue-specific features of the CpG island methylator phenotype
Abstract
Background: The term CpG island methylator phenotype (CIMP) has been used to describe widespread DNA hypermethylation at CpG-rich genomic regions affecting clinically distinct subsets of cancer patients. Even though there have been numerous studies of CIMP in individual cancer types, a uniform analysis across tissues is still lacking.
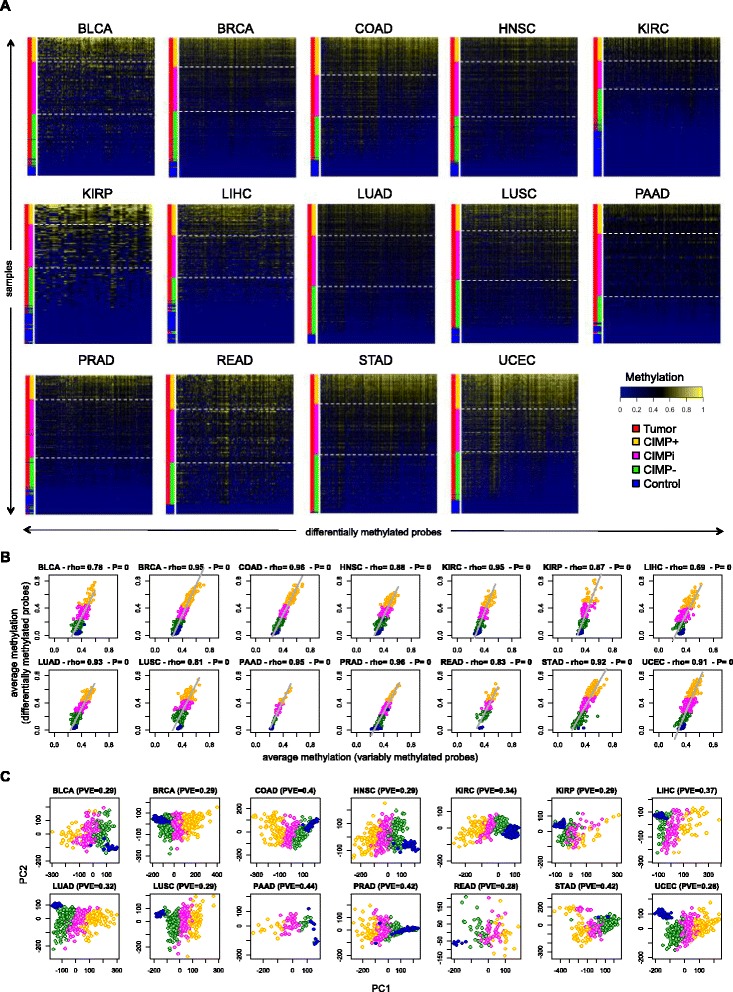
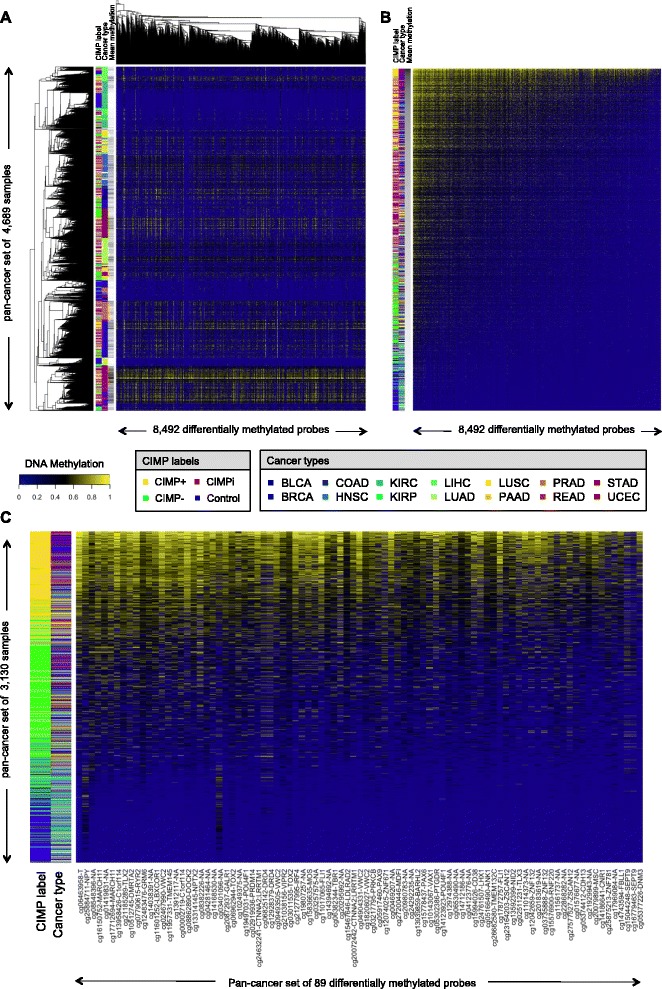
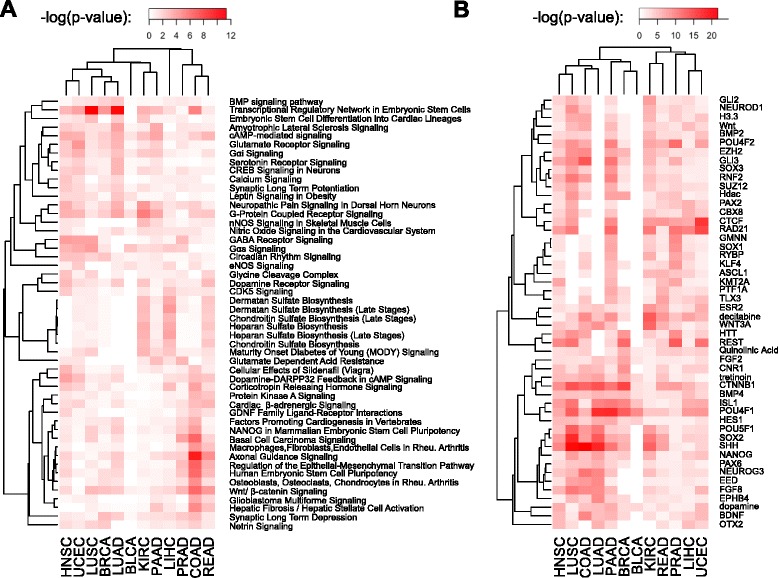
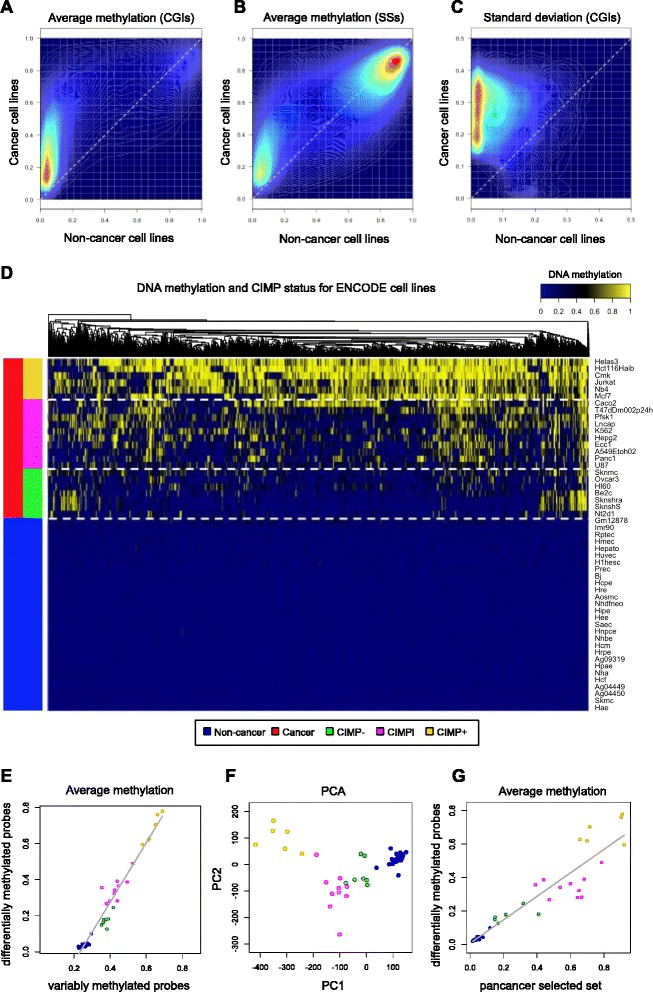
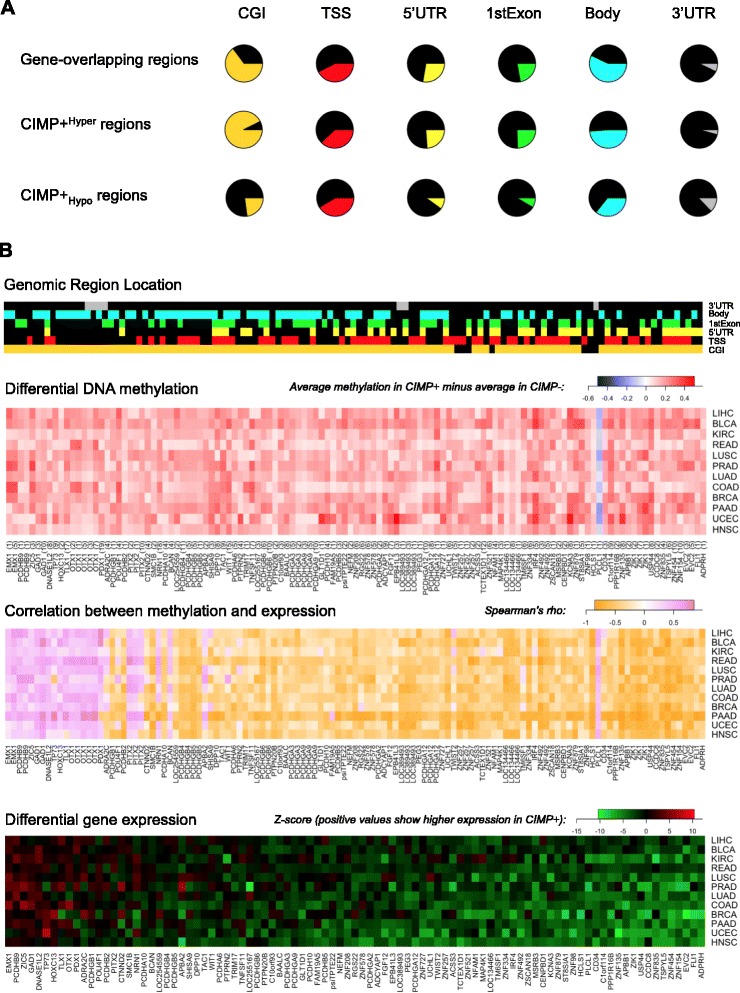
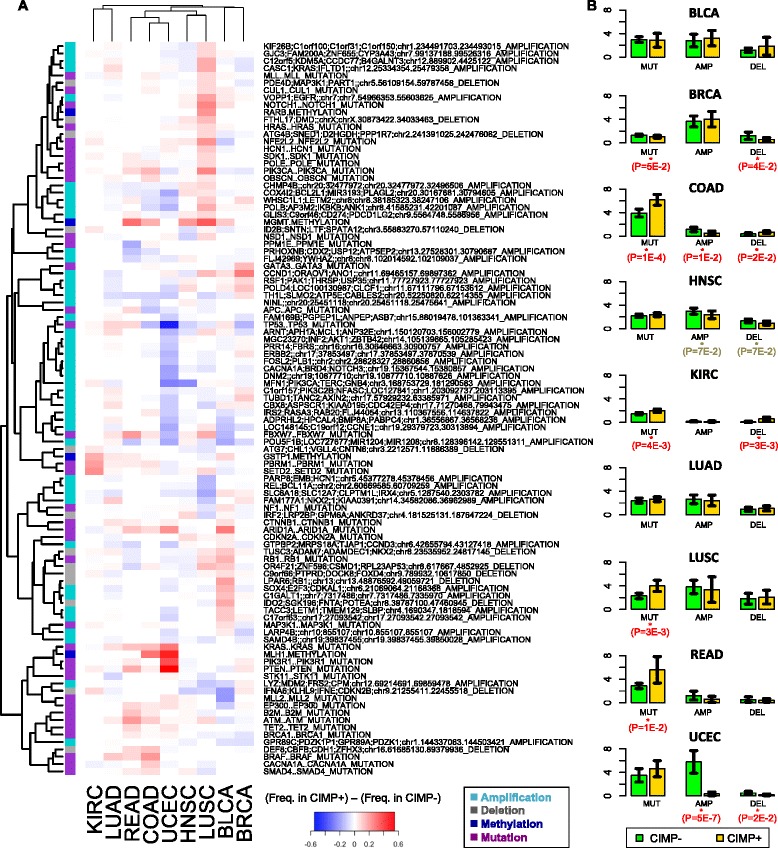
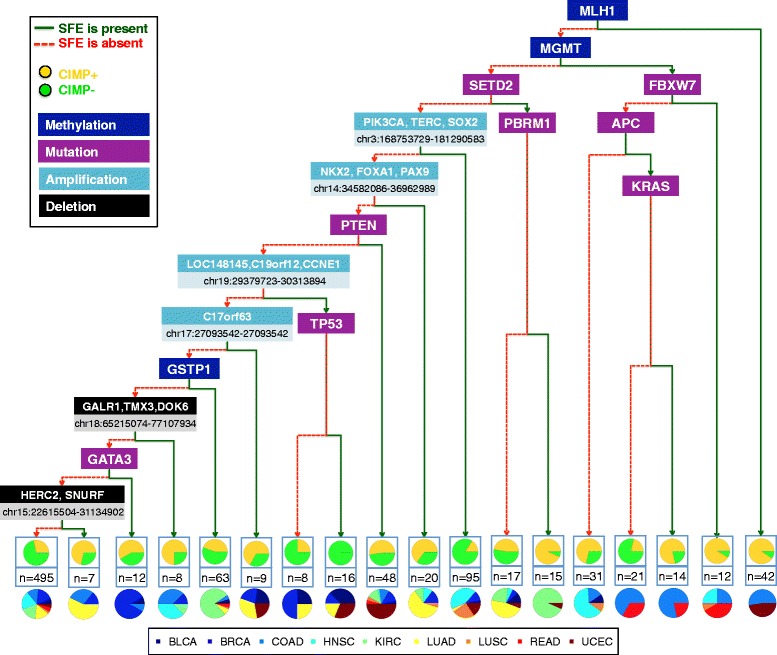
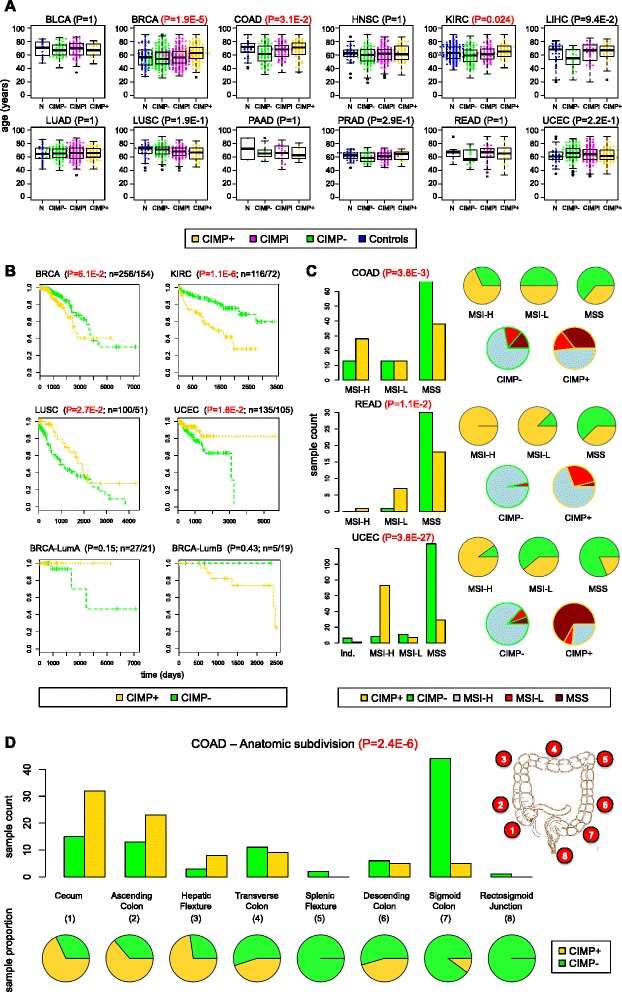
Results: We analyze genome-wide patterns of CpG island hypermethylation in 5,253 solid epithelial tumors from 15 cancer types from TCGA and 23 cancer cell lines from ENCODE. We identify differentially methylated loci that define CIMP+ and CIMP- samples, and we use unsupervised clustering to provide a robust molecular stratification of tumor methylomes for 12 cancer types and all cancer cell lines. With a minimal set of 89 discriminative loci, we demonstrate accurate pan-cancer separation of the 12 CIMP+/- subpopulations, based on their average levels of methylation. Tumor samples in different CIMP subclasses show distinctive correlations with gene expression profiles and recurrence of somatic mutations, copy number variations, and epigenetic silencing. Enrichment analyses indicate shared canonical pathways and upstream regulators for CIMP-targeted regions across cancer types. Furthermore, genomic alterations showing consistent associations with CIMP+/- status include genes involved in DNA repair, chromatin remodeling genes, and several histone methyltransferases. Associations of CIMP status with specific clinical features, including overall survival in several cancer types, highlight the importance of the CIMP+/- designation for individual tumor evaluation and personalized medicine.
Conclusions: We present a comprehensive computational study of CIMP that reveals pan-cancer commonalities and tissue-specific differences underlying concurrent hypermethylation of CpG islands across tumors. Our stratification of solid tumors and cancer cell lines based on CIMP status is data-driven and agnostic to tumor type by design, which protects against known biases that have hindered classic methods previously used to define CIMP. The results that we provide can be used to refine existing molecular subtypes of cancer into more homogeneously behaving subgroups, potentially leading to more uniform responses in clinical trials.
Keywords: CIMP; Cancer; CpG island methylator phenotype; DNA methylation; ENCODE; Pan-cancer; TCGA.
Figures
Similar articles
-
The CpG island methylator phenotype correlates with long-range epigenetic silencing in colorectal cancer.Mol Cancer Res. 2008 Apr;6(4):585-91. doi: 10.1158/1541-7786.MCR-07-2158. Mol Cancer Res. 2008. PMID: 18403637
-
CpG island methylator phenotype in adenocarcinomas from the digestive tract: Methods, conclusions, and controversies.World J Gastrointest Oncol. 2017 Mar 15;9(3):105-120. doi: 10.4251/wjgo.v9.i3.105. World J Gastrointest Oncol. 2017. PMID: 28344746 Free PMC article.
-
Genome-wide methylation analysis identifies a core set of hypermethylated genes in CIMP-H colorectal cancer.BMC Cancer. 2017 Mar 28;17(1):228. doi: 10.1186/s12885-017-3226-4. BMC Cancer. 2017. PMID: 28351398 Free PMC article.
-
The Emergence of Pan-Cancer CIMP and Its Elusive Interpretation.Biomolecules. 2016 Nov 22;6(4):45. doi: 10.3390/biom6040045. Biomolecules. 2016. PMID: 27879658 Free PMC article. Review.
-
Biological significance of the CpG island methylator phenotype.Biochem Biophys Res Commun. 2014 Dec 5;455(1-2):35-42. doi: 10.1016/j.bbrc.2014.07.007. Epub 2014 Jul 10. Biochem Biophys Res Commun. 2014. PMID: 25016183 Review.
Cited by
-
Pan-Cancer Analysis of Human Kinome Gene Expression and Promoter DNA Methylation Identifies Dark Kinase Biomarkers in Multiple Cancers.Cancers (Basel). 2021 Mar 10;13(6):1189. doi: 10.3390/cancers13061189. Cancers (Basel). 2021. PMID: 33801837 Free PMC article.
-
Deciphering the etiology and role in oncogenic transformation of the CpG island methylator phenotype: a pan-cancer analysis.Brief Bioinform. 2022 Mar 10;23(2):bbab610. doi: 10.1093/bib/bbab610. Brief Bioinform. 2022. PMID: 35134107 Free PMC article.
-
Multi-omic signatures identify pan-cancer classes of tumors beyond tissue of origin.Sci Rep. 2020 May 20;10(1):8341. doi: 10.1038/s41598-020-65119-5. Sci Rep. 2020. PMID: 32433524 Free PMC article.
-
DNA Methylation Classes of Stage II and III Primary Melanomas and Their Clinical and Prognostic Significance.JCO Precis Oncol. 2024 Nov;8:e2400375. doi: 10.1200/PO-24-00375. Epub 2024 Nov 7. JCO Precis Oncol. 2024. PMID: 39509669
-
Genes involved in development and differentiation are commonly methylated in cancers derived from multiple organs: a single-institutional methylome analysis using 1007 tissue specimens.Carcinogenesis. 2017 Mar 1;38(3):241-251. doi: 10.1093/carcin/bgw209. Carcinogenesis. 2017. PMID: 28069692 Free PMC article.
References
LinkOut - more resources
Full Text Sources
Other Literature Sources
