

Solution NMR studies of periplasmic binding proteins and their interaction partners
- PMID: 25962019
- PMCID: PMC5506692
- DOI: 10.1515/bmc.2011.005
Solution NMR studies of periplasmic binding proteins and their interaction partners
Abstract
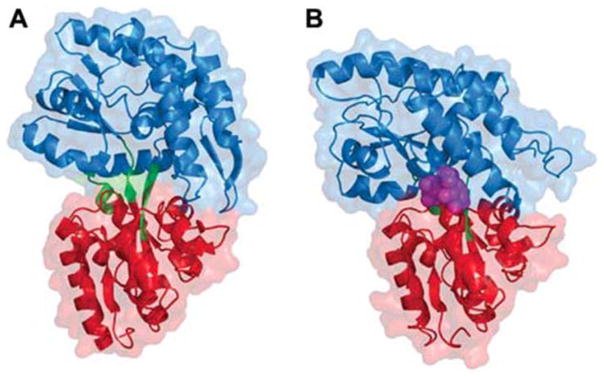
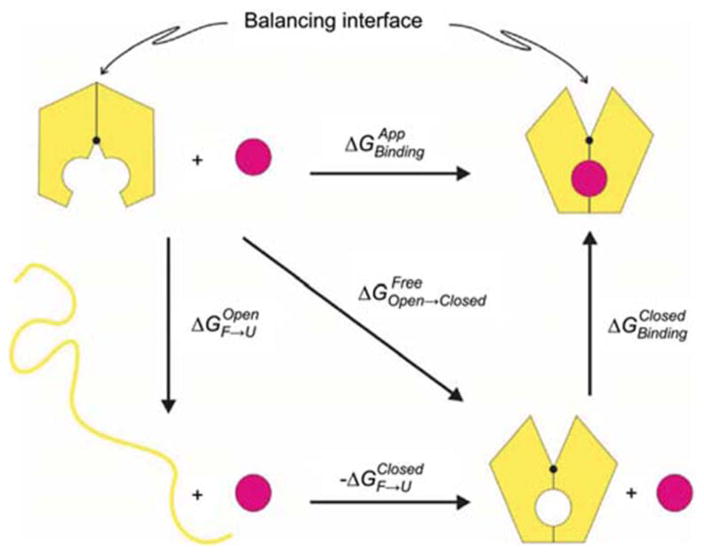
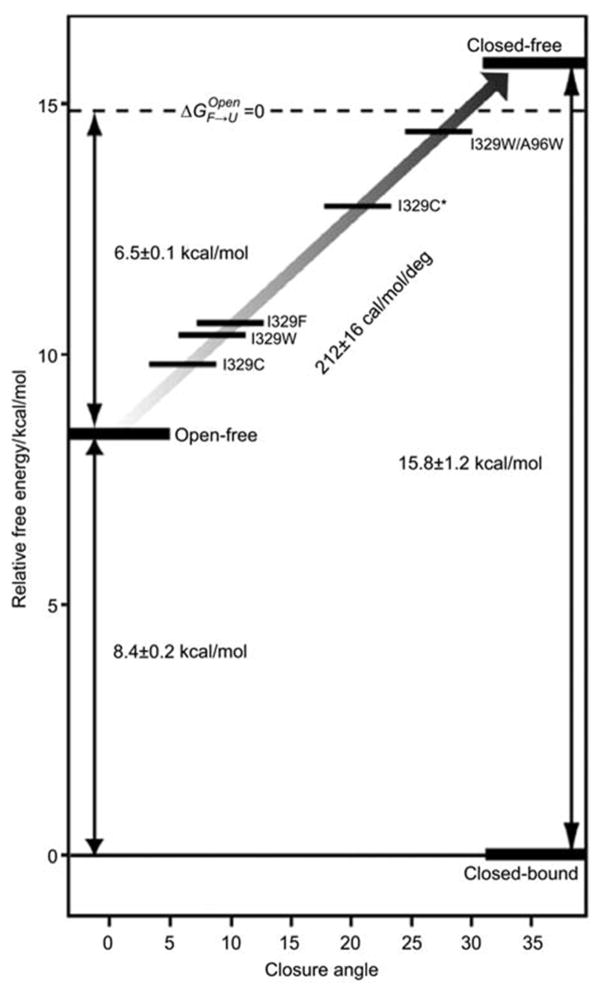
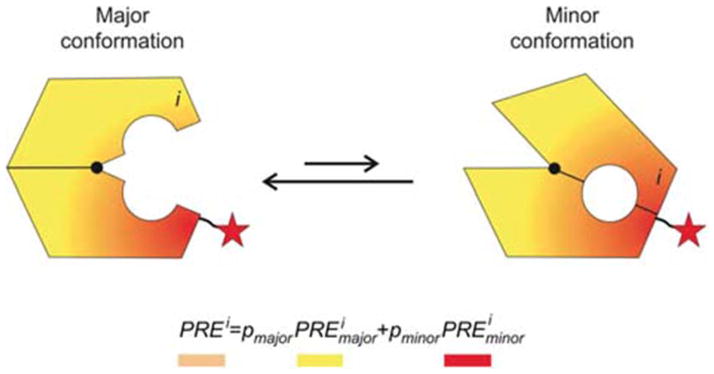
Periplasmic binding proteins (PBPs) are a crucial part of ATP-binding cassette import systems in Gram-negative bacteria. Central to their function is the ability to undergo a large-scale conformational rearrangement from open-unliganded to closed-liganded, which signals the presence of substrate and starts its translocation. Over the years, PBPs have been extensively studied not only owing to their essential role in nutrient uptake but also because they serve as excellent models for both practical applications (e.g., biosensor technology) and basic research (e.g., allosteric mechanisms). Although much of our knowledge at atomic level has been inferred from the detailed, static pictures afforded by crystallographic studies, nuclear magnetic resonance (NMR) has been able to fill certain gaps in such body of work, particularly with regard to dynamic processes. Here, we review NMR studies on PBPs, and their unique insights on conformation, dynamics, energetics, substrate binding, and interactions with related transport proteins. Based on the analysis of recent paramagnetic NMR results, as well as crystallographic and functional observations, we propose a mechanism that could explain the ability of certain PBPs to achieve a closed conformation in absence of ligand while others seem to remain open until ligand-mediated closure.
Figures
Similar articles
-
Ligand-free open-closed transitions of periplasmic binding proteins: the case of glutamine-binding protein.Biochemistry. 2010 Mar 9;49(9):1893-902. doi: 10.1021/bi902045p. Biochemistry. 2010. PMID: 20141110 Free PMC article.
-
Accessing a hidden conformation of the maltose binding protein using accelerated molecular dynamics.PLoS Comput Biol. 2011 Apr;7(4):e1002034. doi: 10.1371/journal.pcbi.1002034. Epub 2011 Apr 21. PLoS Comput Biol. 2011. PMID: 21533070 Free PMC article.
-
Trapping open and closed forms of FitE: a group III periplasmic binding protein.Proteins. 2009 May 15;75(3):598-609. doi: 10.1002/prot.22272. Proteins. 2009. PMID: 19004000
-
Enzyme dynamics from NMR spectroscopy.Acc Chem Res. 2015 Feb 17;48(2):457-65. doi: 10.1021/ar500340a. Epub 2015 Jan 9. Acc Chem Res. 2015. PMID: 25574774 Free PMC article. Review.
-
The Venus flytrap of periplasmic binding proteins: an ancient protein module present in multiple drug receptors.AAPS PharmSci. 1999;1(2):E2. doi: 10.1208/ps010202. AAPS PharmSci. 1999. PMID: 11741199 Free PMC article. Review.
Cited by
-
Temperature dependence of molecular interactions involved in defining stability of glutamine binding protein and its complex with L-glutamine.Biochemistry. 2012 Jan 17;51(2):643-52. doi: 10.1021/bi201494h. Epub 2012 Jan 6. Biochemistry. 2012. PMID: 22206385 Free PMC article.
-
Engineering a Conformationally Switchable Artificial Metalloprotein.J Am Chem Soc. 2022 Nov 30;144(47):21606-21616. doi: 10.1021/jacs.2c08885. Epub 2022 Nov 15. J Am Chem Soc. 2022. PMID: 36378237 Free PMC article.
-
NMR Analysis of Apo Glutamine-Binding Protein Exposes Challenges in the Study of Interdomain Dynamics.Angew Chem Int Ed Engl. 2019 Nov 18;58(47):16899-16902. doi: 10.1002/anie.201911015. Epub 2019 Oct 11. Angew Chem Int Ed Engl. 2019. PMID: 31515908 Free PMC article.
-
Conformation-Dependent Hydrogen-Bonding Interactions in a Switchable Artificial Metalloprotein.Biochemistry. 2024 Aug 20;63(16):2040-2050. doi: 10.1021/acs.biochem.4c00209. Epub 2024 Aug 1. Biochemistry. 2024. PMID: 39088332 Free PMC article.
-
Structure-driven development of a biomimetic rare earth artificial metalloprotein.Proc Natl Acad Sci U S A. 2024 Aug 13;121(33):e2405836121. doi: 10.1073/pnas.2405836121. Epub 2024 Aug 8. Proc Natl Acad Sci U S A. 2024. PMID: 39116128 Free PMC article.
References
-
- Holland IB, Cole SPC, Kuchler K, Higgins CF, editors. ABC proteins: from bacteria to man. 1. Amsterdam/Boston, MA: Academic Press; 2003.
-
- Quiocho FA. Atomic structures of periplasmic binding-proteins and the high-affinity active-transport systems in bacteria. Philos Trans R Soc Lond B. 1990;326:341–52. - PubMed
-
- Quiocho FA. Atomic structures and function of periplasmic receptors for active transport and chemotaxis. Curr Opin Struct Biol. 1991;1:922–33.
-
- Quiocho FA, Ledvina PS. Atomic structure and specificity of bacterial periplasmic receptors for active transport and chemotaxis: variation of common themes. Mol Microbiol. 1996;20:17–25. - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources