

Genetic testing of Korean familial hypercholesterolemia using whole-exome sequencing
- PMID: 25962062
- PMCID: PMC4427254
- DOI: 10.1371/journal.pone.0126706
Genetic testing of Korean familial hypercholesterolemia using whole-exome sequencing
Abstract
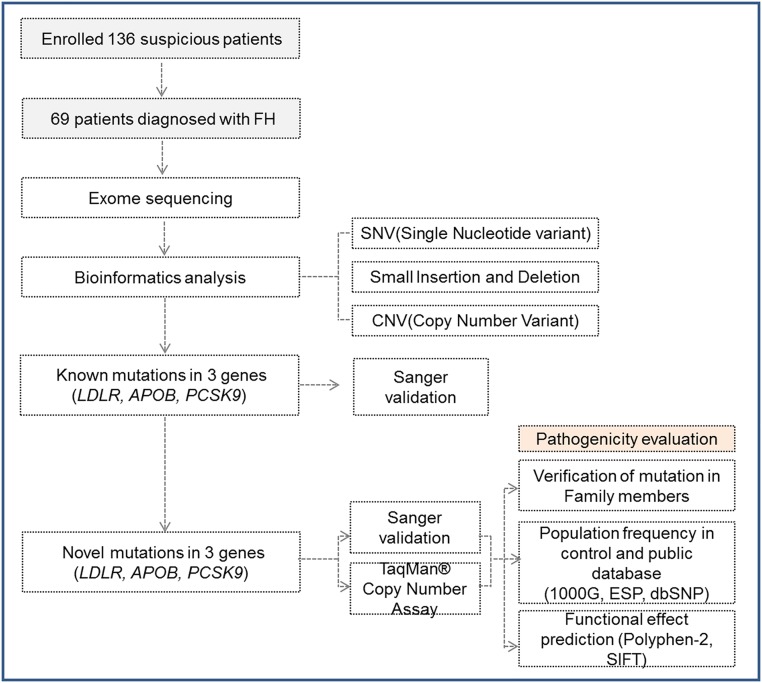
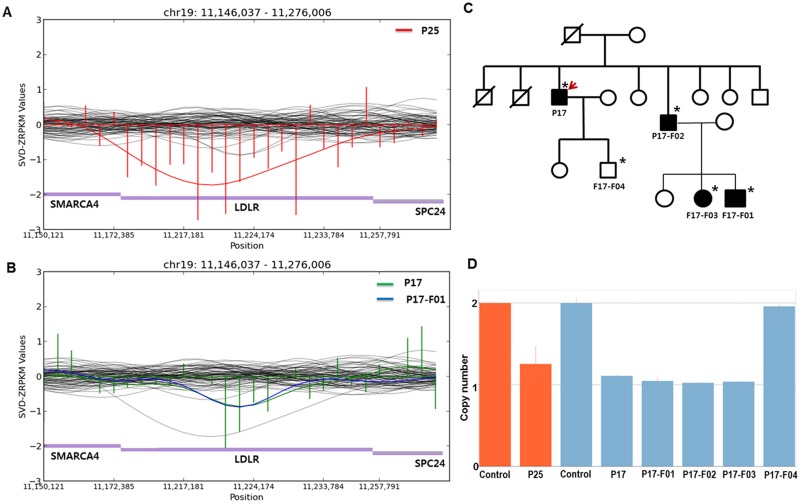
Familial hypercholesterolemia (FH) is a genetic disorder with an increased risk of early-onset coronary artery disease. Although some clinically diagnosed FH cases are caused by mutations in LDLR, APOB, or PCSK9, mutation detection rates and profiles can vary across ethnic groups. In this study, we aimed to provide insight into the spectrum of FH-causing mutations in Koreans. Among 136 patients referred for FH, 69 who met Simon Broome criteria with definite family history were enrolled. By whole-exome sequencing (WES) analysis, we confirmed that the 3 known FH-related genes accounted for genetic causes in 23 patients (33.3%). A substantial portion of the mutations (19 of 23 patients, 82.6%) resulted from 17 mutations and 2 copy number deletions in LDLR gene. Two mutations each in the APOB and PCSK9 genes were verified. Of these anomalies, two frameshift deletions in LDLR and one mutation in PCSK9 were identified as novel causative mutations. In particular, one novel mutation and copy number deletion were validated by co-segregation in their relatives. This study confirmed the utility of genetic diagnosis of FH through WES.
Conflict of interest statement
Figures

References
-
- Sharma P, Boyers D, Boachie C, Stewart F, Miedzybrodzka Z, Simpson W, et al. Elucigene FH20 and LIPOchip for the diagnosis of familial hypercholesterolaemia: a systematic review and economic evaluation. Health technology assessment (Winchester, England). 2012;16(17):1–266. Epub 2012/04/04. 10.3310/hta16170 . - DOI - PMC - PubMed
-
- Usifo E, Leigh SE, Whittall RA, Lench N, Taylor A, Yeats C, et al. Low-density lipoprotein receptor gene familial hypercholesterolemia variant database: update and pathological assessment. Annals of human genetics. 2012;76(5):387–401. Epub 2012/08/14. 10.1111/j.1469-1809.2012.00724.x . - DOI - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
