

Estimating the mutation load in human genomes
- PMID: 25963372
- PMCID: PMC4959039
- DOI: 10.1038/nrg3931
Estimating the mutation load in human genomes
Abstract
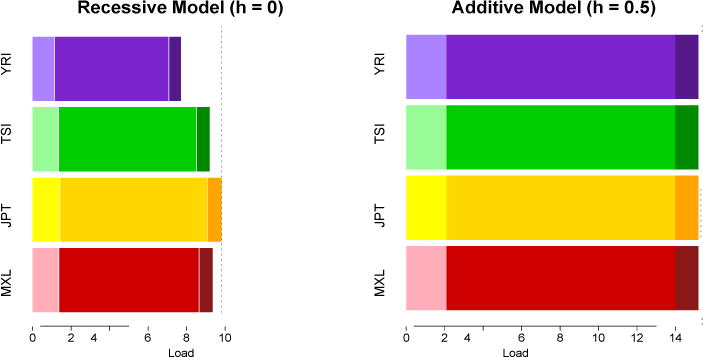
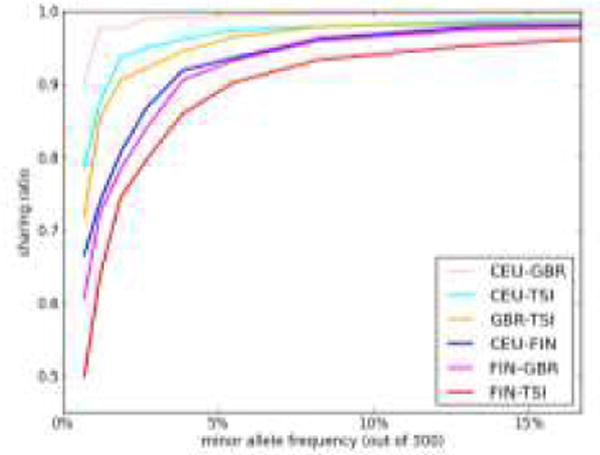
Next-generation sequencing technology has facilitated the discovery of millions of genetic variants in human genomes. A sizeable fraction of these variants are predicted to be deleterious. Here, we review the pattern of deleterious alleles as ascertained in genome sequencing data sets and ask whether human populations differ in their predicted burden of deleterious alleles - a phenomenon known as mutation load. We discuss three demographic models that are predicted to affect mutation load and relate these models to the evidence (or the lack thereof) for variation in the efficacy of purifying selection in diverse human genomes. We also emphasize why accurate estimation of mutation load depends on assumptions regarding the distribution of dominance and selection coefficients - quantities that remain poorly characterized for current genomic data sets.
Figures
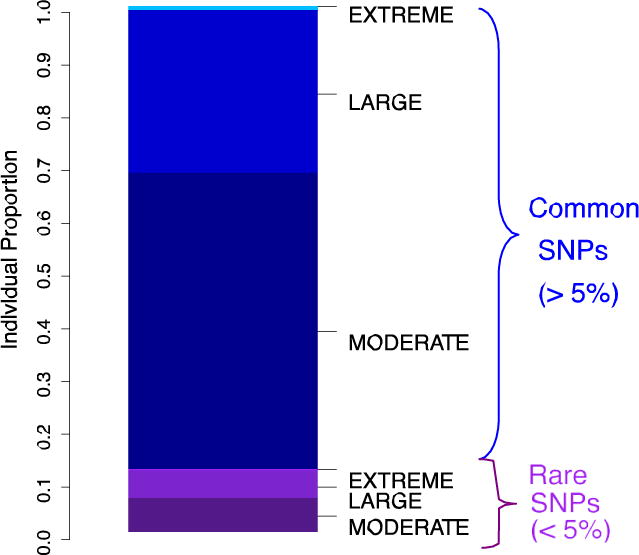
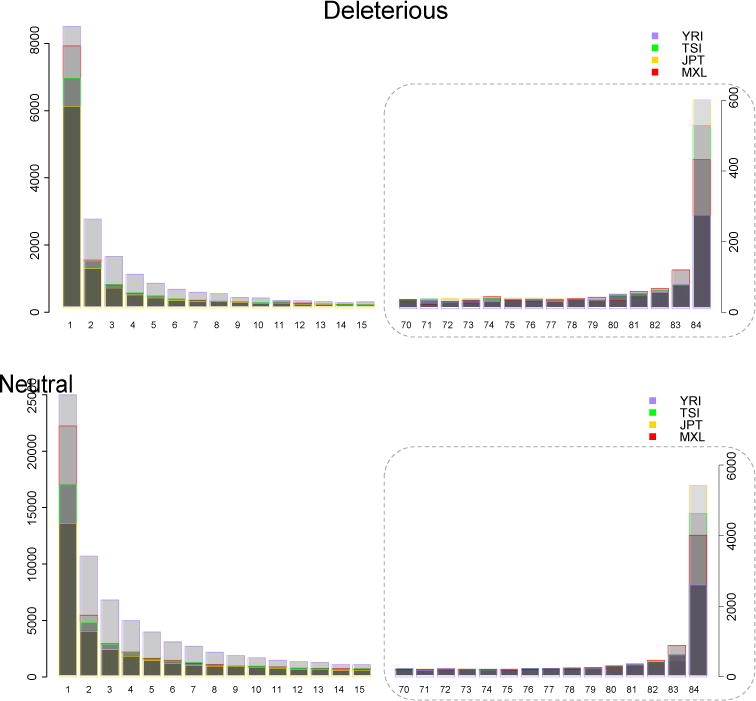
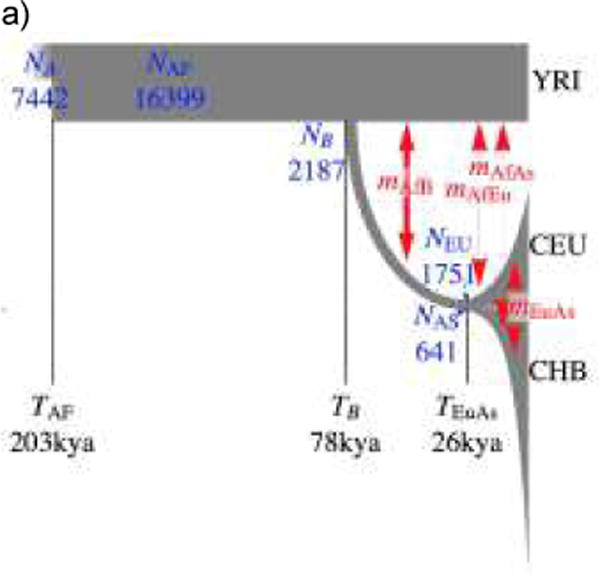
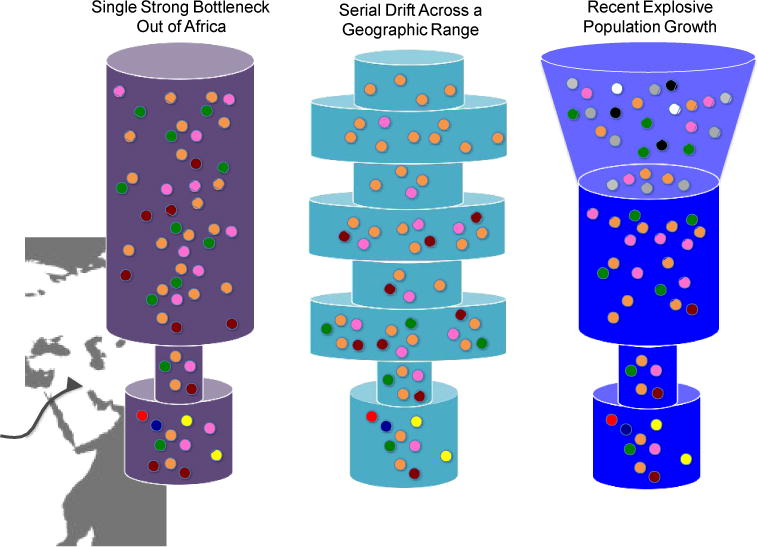
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
