

Choice of Starting Dose for Biopharmaceuticals in First-in-Human Phase I Cancer Clinical Trials
- PMID: 25964306
- PMCID: PMC4571794
- DOI: 10.1634/theoncologist.2015-0008
Choice of Starting Dose for Biopharmaceuticals in First-in-Human Phase I Cancer Clinical Trials
Abstract
Background: First-in-human (FIH) trials of low-molecular-weight anticancer agents conventionally derive a safe start dose (SD) from one-tenth the severely toxic dose in 10% of rodents or one-sixth the highest nonseverely toxic dose (HNSTD) in nonrodent species. No consensus has been reached on whether this paradigm can be safely applied to biotechnology-derived products (BDPs).
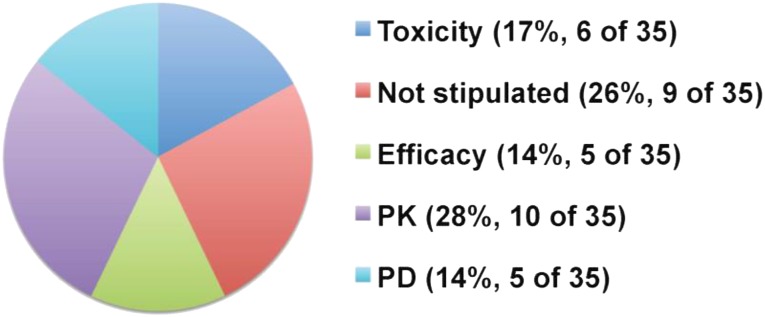
Materials and methods: A comprehensive search was conducted to identify all BDPs (excluding immune checkpoint inhibitors and antibody drug conjugates) with sufficient nonclinical and clinical data to assess the safety of hypothetical use of one-sixth HNSTD in an advanced cancer FIH trial.
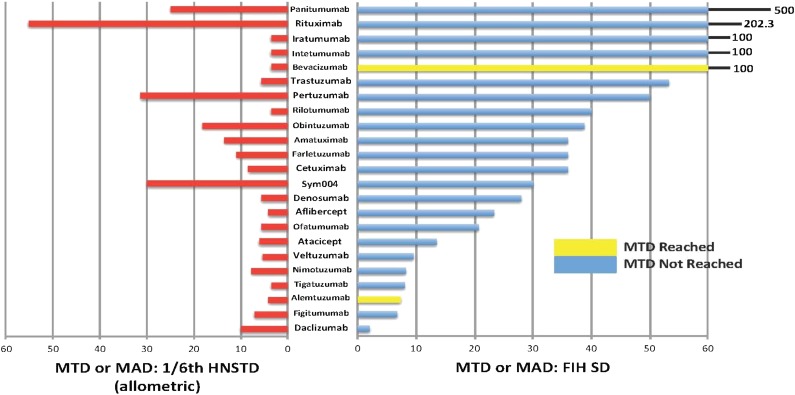
Results: The search identified 23 BDPs, of which 21 were monoclonal antibodies. The median ratio of the maximum tolerated or maximum administered dose (MTD or MAD) to the actual FIH SD was 36 (range, 8-500). Only 2 BDPs reached the MTD. Hypothetical use of one-sixth HNSTD (allometrically scaled to humans) would not have exceeded the MTD or MAD for all 23 BDPs and would have reduced the median ratio of the MTD or MAD to a SD to 6.1 (range, 3.5-55.3). Pharmacodynamic (PD) markers were included in some animal toxicology studies and were useful to confirm the hypothetical SD of one-sixth HNSTD.
Conclusion: One-sixth HNSTD would not have resulted in unacceptable toxicities in the data available. Supporting its use could reduce the number of dose escalations needed to reach the recommended dose. A low incidence of toxicities in animals and humans underscores the need to identify the pharmacokinetic and PD parameters to guide SD selection of BDPs for FIH cancer trials.
Implications for practice: Start dose (SD) for biotechnology-derived products (BDPs) can be safely derived from one-sixth the highest nonseverely toxic dose in nonrodent species and may reduce the number of dose escalations needed to reach the recommended dose in first-in-human studies while limiting unnecessary exposure to high drug levels in humans. The use of this type of SD could improve the design of phase I studies of BDPs by making them more efficient. The role of preclinical pharmacodynamic markers was useful in confirming the hypothetical SD, and attempts should be explored in future animal studies to identify such parameters.
Keywords: Animal toxicology studies; Biotechnology derived products; Highest nonseverely toxic dose; Phase I clinical trials; Starting dose.
©AlphaMed Press.
Conflict of interest statement
Disclosures of potential conflicts of interest may be found at the end of this article.
Figures
References
-
- Schein PS. Preclinical toxicology of anticancer agents. Cancer Res. 1977;37:1934–1937. - PubMed
-
- International Conference for Harmonisation. S9 Nonclinical Evaluation for Anticancer Pharmaceuticals 2010. Available at http://www.fda.gov/ucm/groups/fdagov-public/@fdagov-drugs-gen/documents/.... Accessed January 5, 2015.
-
- Rozencweig M, Von Hoff DD, Staquet MJ, et al. Animal toxicology for early clinical trials with anticancer agents. Cancer Clin Trials. 1981;4:21–28. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
