

Conserved RNA secondary structures and long-range interactions in hepatitis C viruses
- PMID: 25964384
- PMCID: PMC4478341
- DOI: 10.1261/rna.049338.114
Conserved RNA secondary structures and long-range interactions in hepatitis C viruses
Erratum in
-
Corrigendum: Conserved RNA secondary structures and long-range interactions in hepatitis C viruses.RNA. 2016 Oct;22(10):1640.1. doi: 10.1261/rna.058123.116. RNA. 2016. PMID: 27638914 Free PMC article. No abstract available.
Abstract
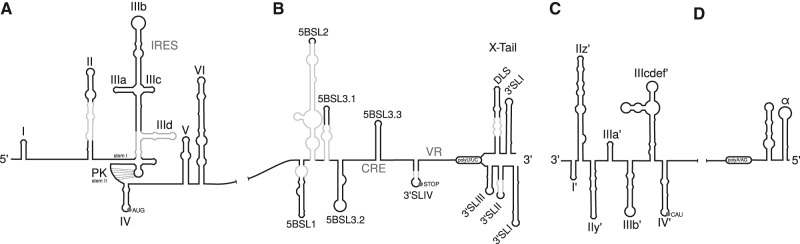
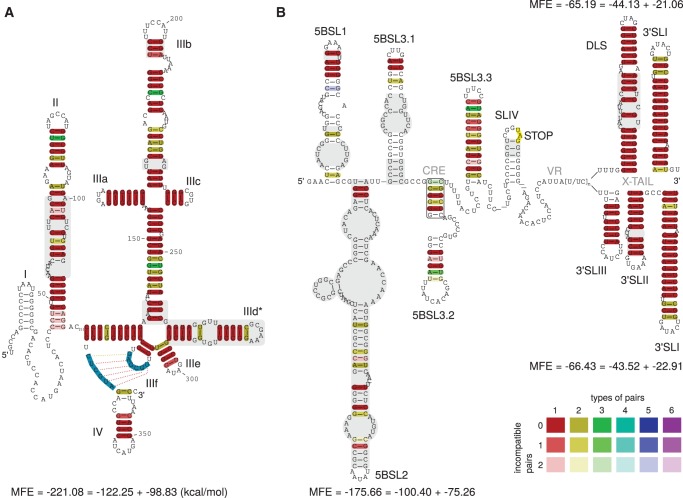
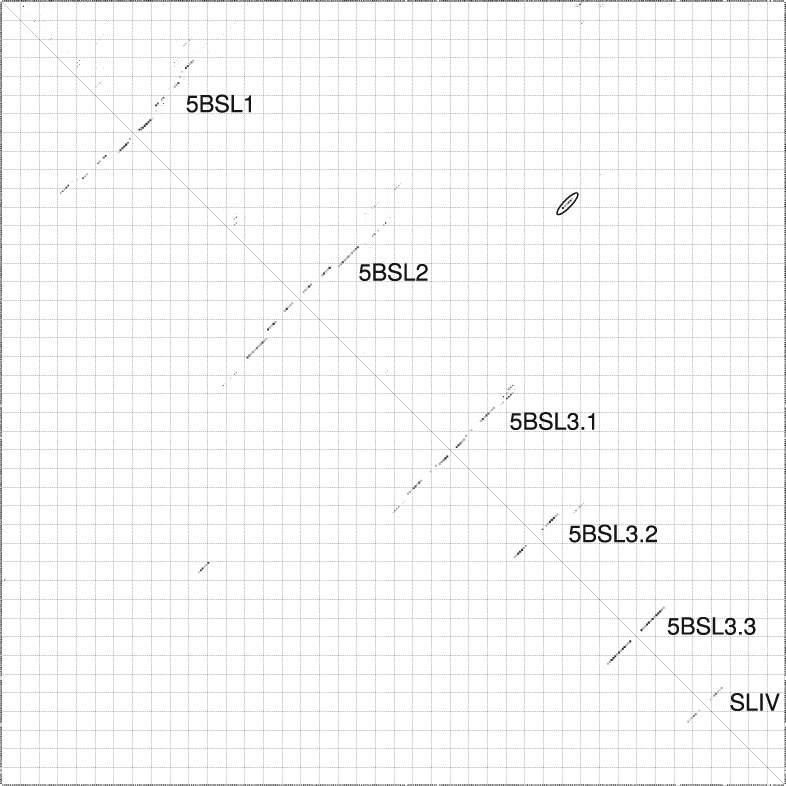
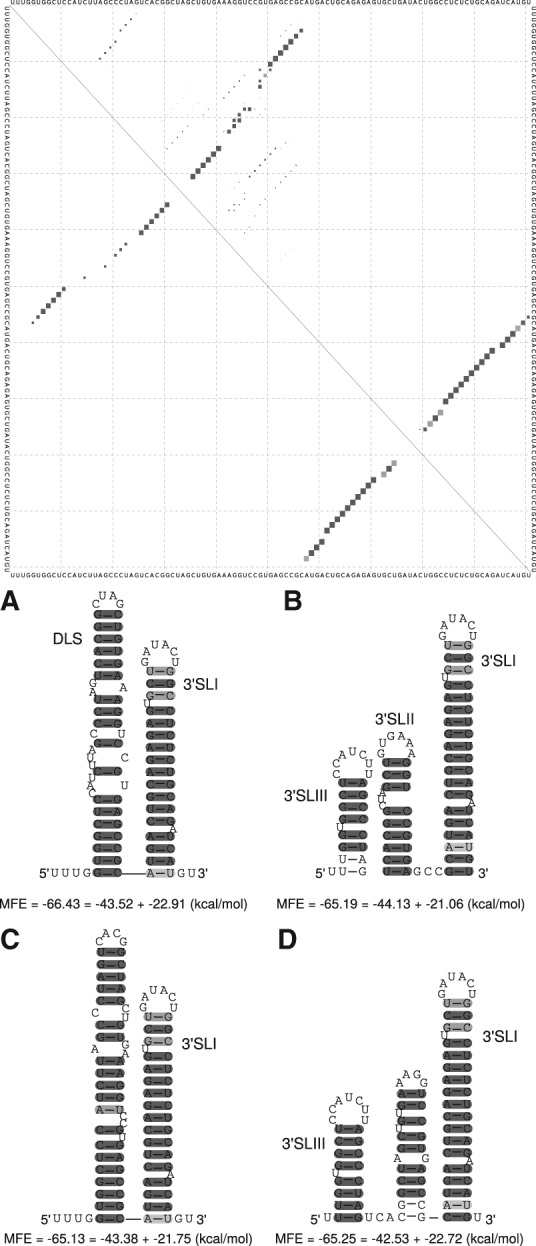
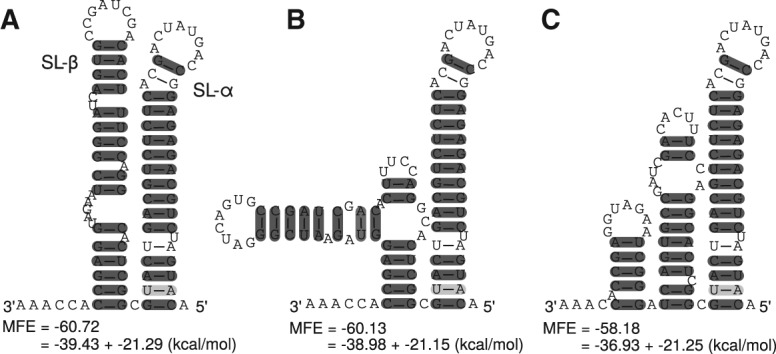
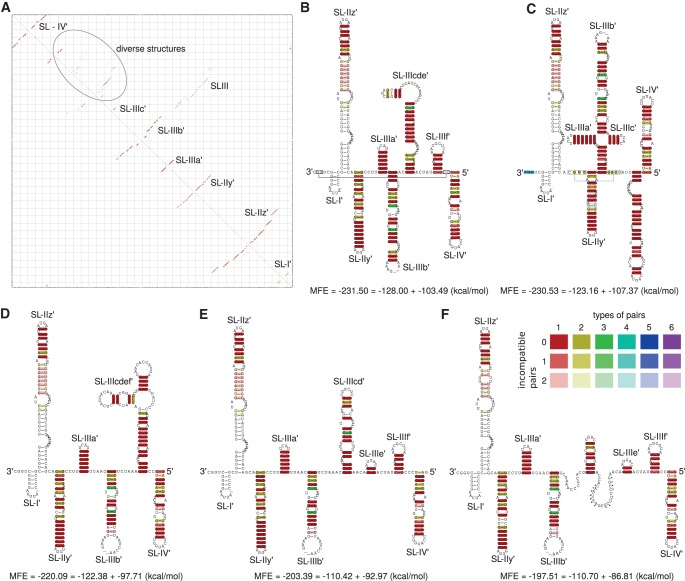
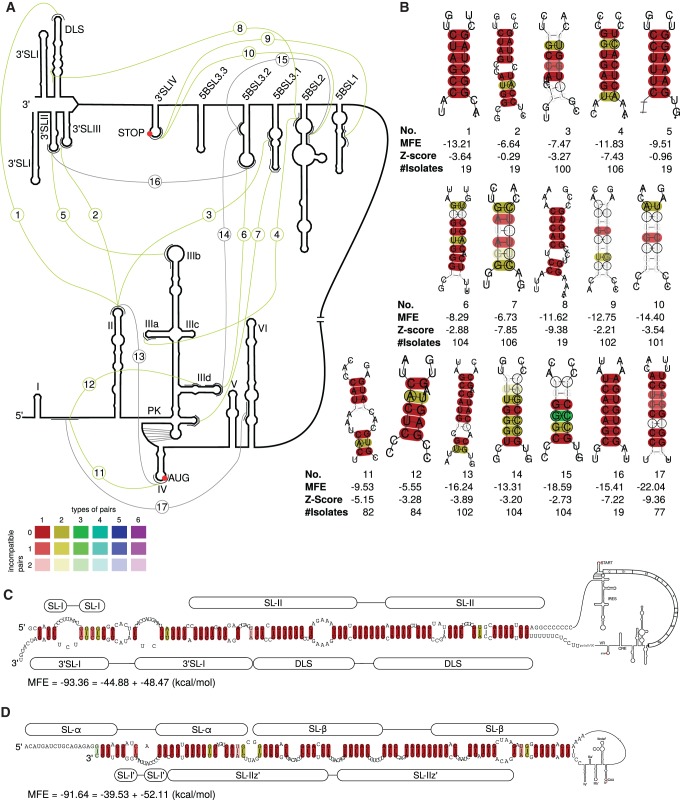
Hepatitis C virus (HCV) is a hepatotropic virus with a plus-strand RNA genome of ∼9.600 nt. Due to error-prone replication by its RNA-dependent RNA polymerase (RdRp) residing in nonstructural protein 5B (NS5B), HCV isolates are grouped into seven genotypes with several subtypes. By using whole-genome sequences of 106 HCV isolates and secondary structure alignments of the plus-strand genome and its minus-strand replication intermediate, we established refined secondary structures of the 5' untranslated region (UTR), the cis-acting replication element (CRE) in NS5B, and the 3' UTR. We propose an alternative structure in the 5' UTR, conserved secondary structures of 5B stem-loop (SL)1 and 5BSL2, and four possible structures of the X-tail at the very 3' end of the HCV genome. We predict several previously unknown long-range interactions, most importantly a possible circularization interaction between distinct elements in the 5' and 3' UTR, reminiscent of the cyclization elements of the related flaviviruses. Based on analogy to these viruses, we propose that the 5'-3' UTR base-pairing in the HCV genome might play an important role in viral RNA replication. These results may have important implications for our understanding of the nature of the cis-acting RNA elements in the HCV genome and their possible role in regulating the mutually exclusive processes of viral RNA translation and replication.
Keywords: HCV; RNA long-range interaction prediction; RNA secondary structure prediction; bioinformatics; circularization.
© 2015 Fricke et al.; Published by Cold Spring Harbor Laboratory Press for the RNA Society.
Figures
Similar articles
-
A hepatitis C virus cis-acting replication element forms a long-range RNA-RNA interaction with upstream RNA sequences in NS5B.J Virol. 2008 Sep;82(18):9008-22. doi: 10.1128/JVI.02326-07. Epub 2008 Jul 9. J Virol. 2008. PMID: 18614633 Free PMC article.
-
microRNA-122 target sites in the hepatitis C virus RNA NS5B coding region and 3' untranslated region: function in replication and influence of RNA secondary structure.Cell Mol Life Sci. 2017 Feb;74(4):747-760. doi: 10.1007/s00018-016-2377-9. Epub 2016 Sep 27. Cell Mol Life Sci. 2017. PMID: 27677491 Free PMC article.
-
Genomic-Scale Interaction Involving Complementary Sequences in the Hepatitis C Virus 5'UTR Domain IIa and the RNA-Dependent RNA Polymerase Coding Region Promotes Efficient Virus Replication.Viruses. 2018 Dec 28;11(1):17. doi: 10.3390/v11010017. Viruses. 2018. PMID: 30597844 Free PMC article.
-
cis-Acting RNA elements in the hepatitis C virus RNA genome.Virus Res. 2015 Aug 3;206:90-8. doi: 10.1016/j.virusres.2014.12.029. Epub 2015 Jan 7. Virus Res. 2015. PMID: 25576644 Free PMC article. Review.
-
Genome cyclization as strategy for flavivirus RNA replication.Virus Res. 2009 Feb;139(2):230-9. doi: 10.1016/j.virusres.2008.07.016. Epub 2008 Sep 9. Virus Res. 2009. PMID: 18703097 Free PMC article. Review.
Cited by
-
Deconstructing internal ribosome entry site elements: an update of structural motifs and functional divergences.Open Biol. 2018 Nov 28;8(11):180155. doi: 10.1098/rsob.180155. Open Biol. 2018. PMID: 30487301 Free PMC article. Review.
-
A small stem-loop structure of the Ebola virus trailer is essential for replication and interacts with heat-shock protein A8.Nucleic Acids Res. 2016 Nov 16;44(20):9831-9846. doi: 10.1093/nar/gkw825. Epub 2016 Sep 19. Nucleic Acids Res. 2016. PMID: 27651462 Free PMC article.
-
Comprehensive survey of conserved RNA secondary structures in full-genome alignment of Hepatitis C virus.Sci Rep. 2024 Jul 2;14(1):15145. doi: 10.1038/s41598-024-62897-0. Sci Rep. 2024. PMID: 38956134 Free PMC article.
-
Distinct roles for the IIId2 sub-domain in pestivirus and picornavirus internal ribosome entry sites.Nucleic Acids Res. 2017 Dec 15;45(22):13016-13028. doi: 10.1093/nar/gkx991. Nucleic Acids Res. 2017. PMID: 29069411 Free PMC article.
-
Women in the European Virus Bioinformatics Center.Viruses. 2022 Jul 12;14(7):1522. doi: 10.3390/v14071522. Viruses. 2022. PMID: 35891501 Free PMC article.
References
-
- Alvarez DE, Filomatori CV, Gamarnik AV. 2008. Functional analysis of dengue virus cyclization sequences located at the 5′ and 3′UTRs. Virology 375: 223–235. - PubMed
-
- Anandam P, Torarinsson E, Ruzzo WL. 2009. Multiperm: shuffling multiple sequence alignments while approximately preserving dinucleotide frequencies. Bioinformatics 25: 668–669. - PubMed
-
- Astier-Gin T, Bellecave P, Litvak S, Ventura M. 2005. Template requirements and binding of hepatitis C virus NS5B polymerase during in vitro RNA synthesis from the 3′-end of virus minus-strand RNA. FEBS J 272: 3872–3886. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials