

Molecular mechanisms of human IRE1 activation through dimerization and ligand binding
- PMID: 25968568
- PMCID: PMC4536996
- DOI: 10.18632/oncotarget.3864
Molecular mechanisms of human IRE1 activation through dimerization and ligand binding
Abstract
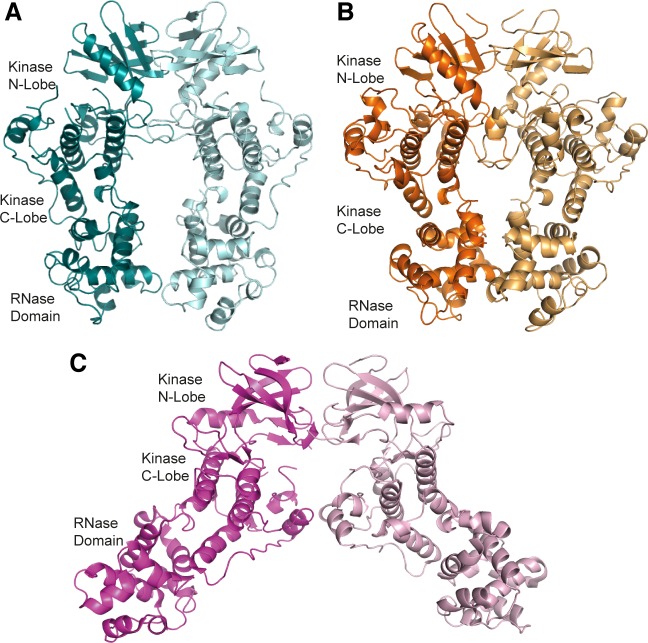
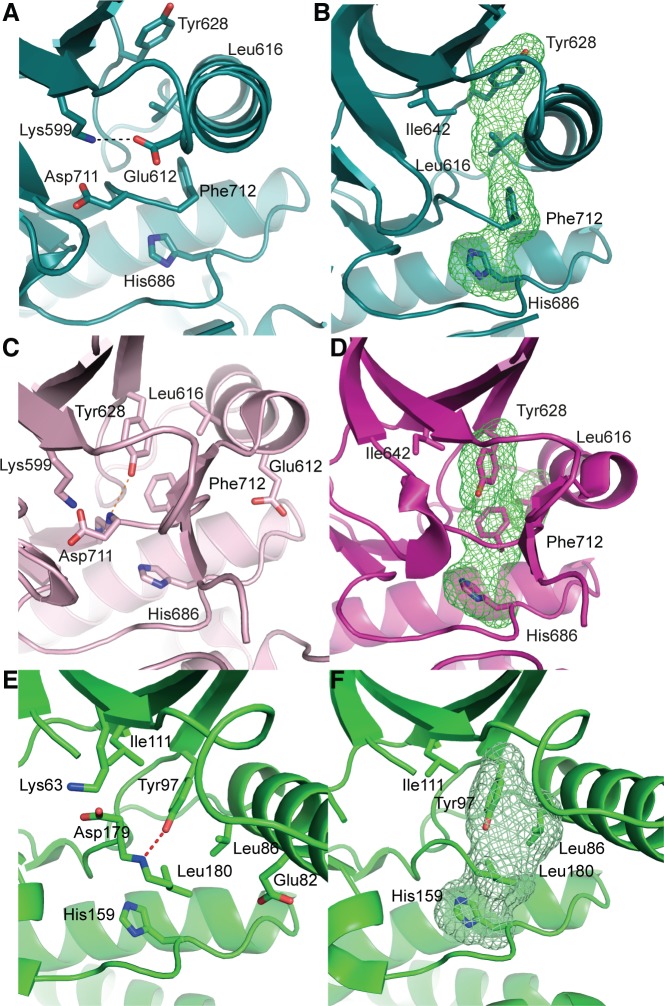
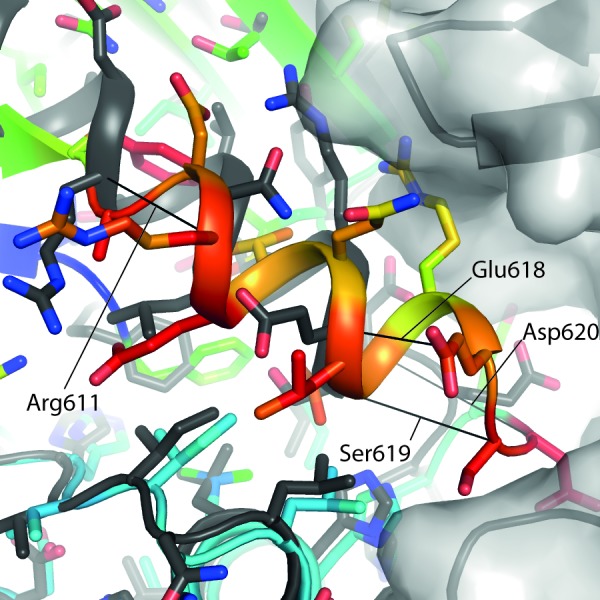
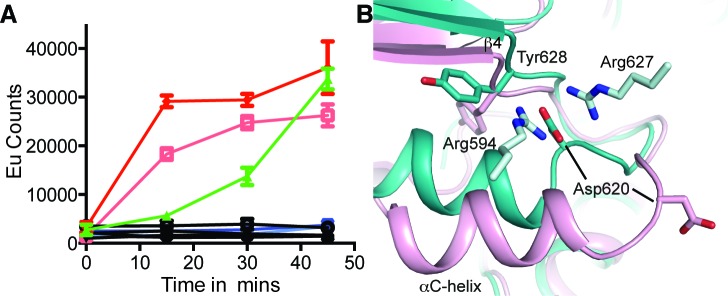
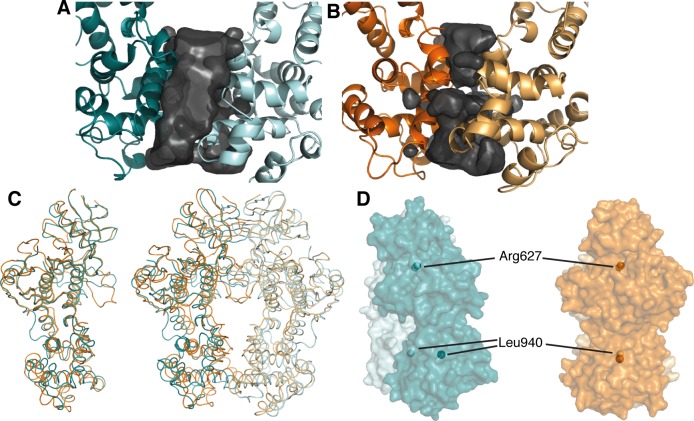
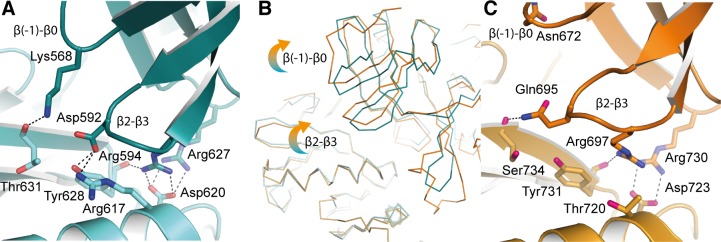
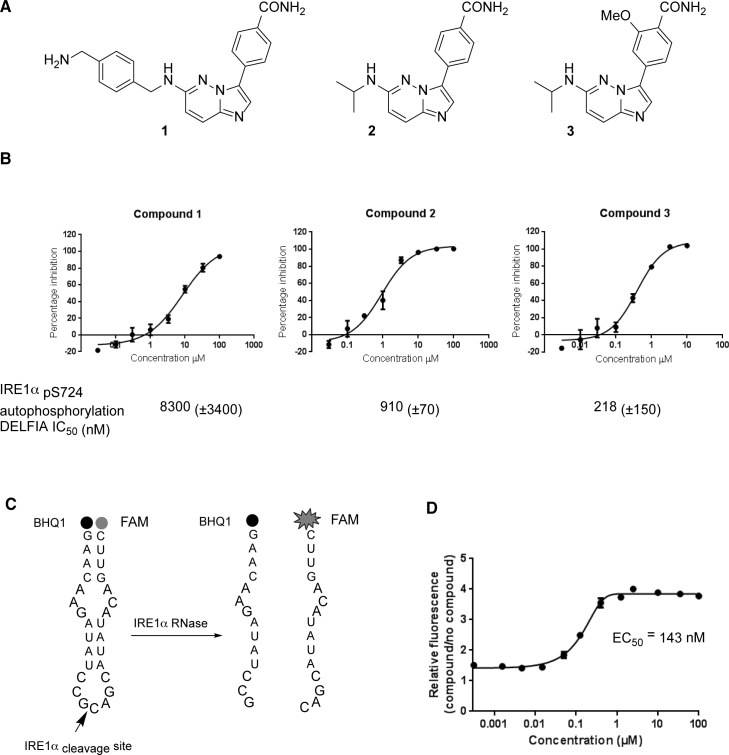
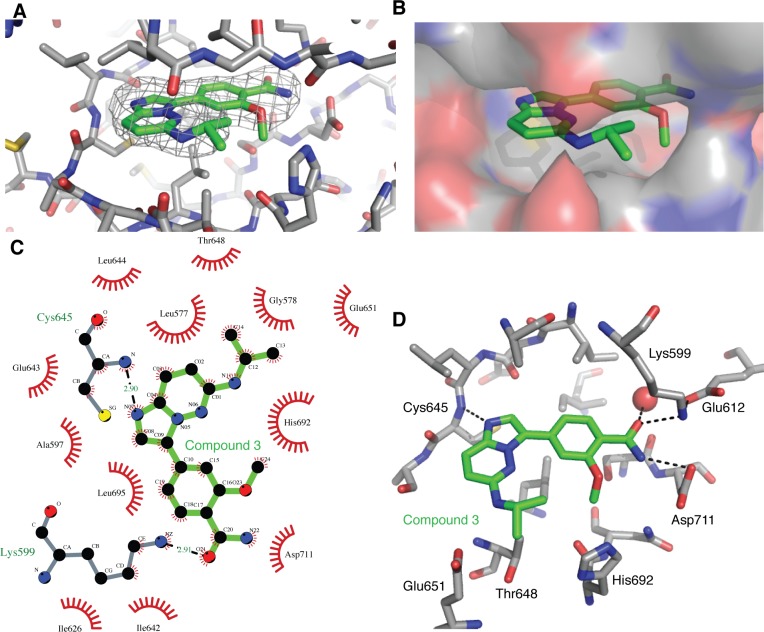
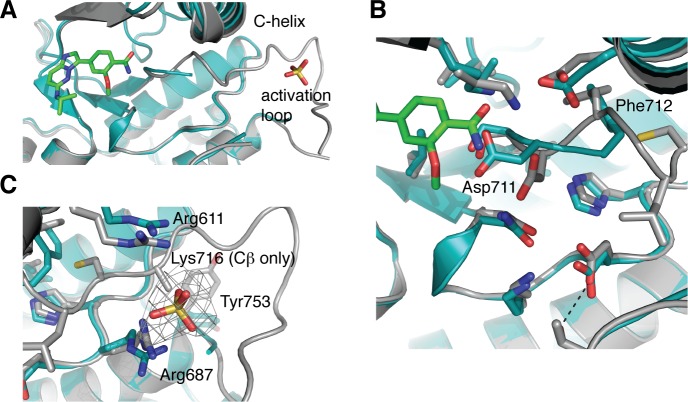
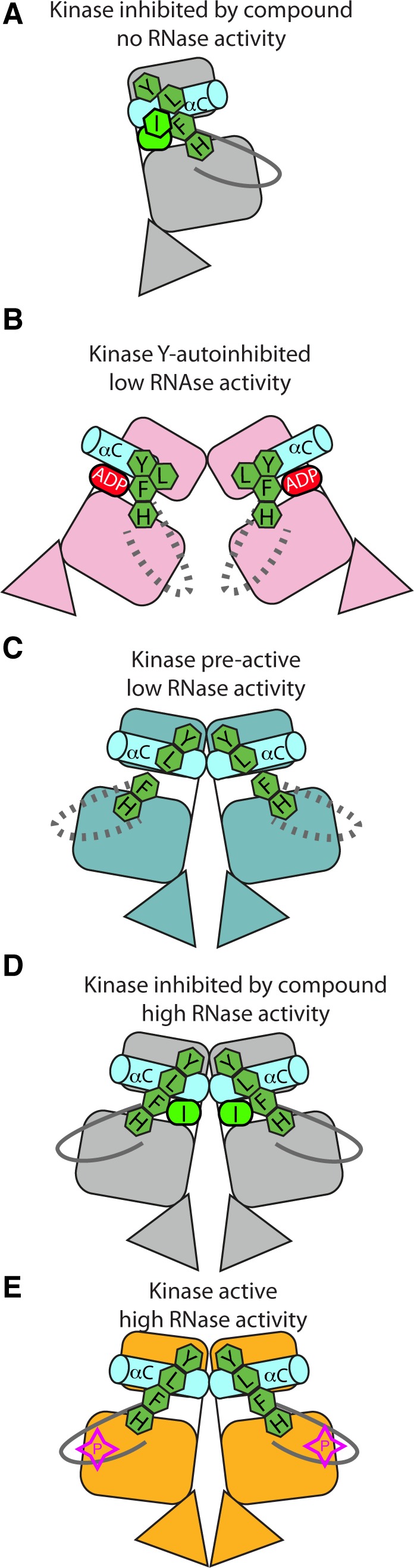
IRE1 transduces the unfolded protein response by splicing XBP1 through its C-terminal cytoplasmic kinase-RNase region. IRE1 autophosphorylation is coupled to RNase activity through formation of a back-to-back dimer, although the conservation of the underlying molecular mechanism is not clear from existing structures. We have crystallized human IRE1 in a back-to-back conformation only previously seen for the yeast homologue. In our structure the kinase domain appears primed for catalysis but the RNase domains are disengaged. Structure-function analysis reveals that IRE1 is autoinhibited through a Tyr-down mechanism related to that found in the unrelated Ser/Thr protein kinase Nek7. We have developed a compound that potently inhibits human IRE1 kinase activity while stimulating XBP1 splicing. A crystal structure of the inhibitor bound to IRE1 shows an increased ordering of the kinase activation loop. The structures of hIRE in apo and ligand-bound forms are consistent with a previously proposed model of IRE1 regulation in which formation of a back-to-back dimer coupled to adoption of a kinase-active conformation drive RNase activation. The structures provide opportunities for structure-guided design of IRE1 inhibitors.
Keywords: RNase; UPR; drug discovery; kinase.
Conflict of interest statement
Please note that all authors who are, or have been, employed by The Institute of Cancer Research are subject to a “Rewards to Inventors Scheme” which may reward contributors to a programme that is subsequently licensed. The Institute of Cancer research has a commercial interest in the development of inhibitors of IRE1.
Figures
References
-
- Walter P., Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science. 2011;334:1081–6. - PubMed
-
- Ron D., Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol. 2007;8:519–29. - PubMed
-
- Sidrauski C., Walter P. The transmembrane kinase Ire1p is a site-specific endonuclease that initiates mRNA splicing in the unfolded protein response. Cell. 1997;90:1031–9. - PubMed
-
- Cox J.S., Shamu C.E., Walter P. Transcriptional induction of genes encoding endoplasmic reticulum resident proteins requires a transmembrane protein kinase. Cell. 1993;73:1197–206. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
