

Rapid quantification of mutant fitness in diverse bacteria by sequencing randomly bar-coded transposons
- PMID: 25968644
- PMCID: PMC4436071
- DOI: 10.1128/mBio.00306-15
Rapid quantification of mutant fitness in diverse bacteria by sequencing randomly bar-coded transposons
Abstract
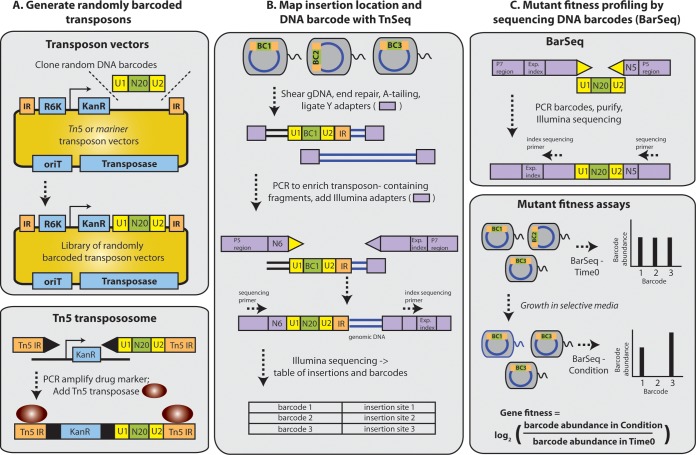
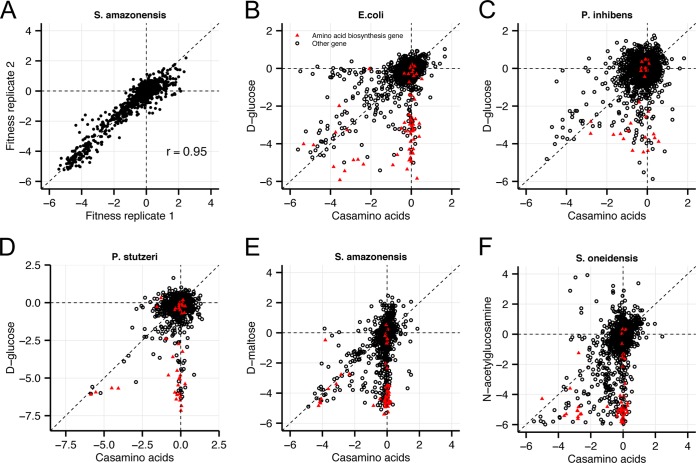
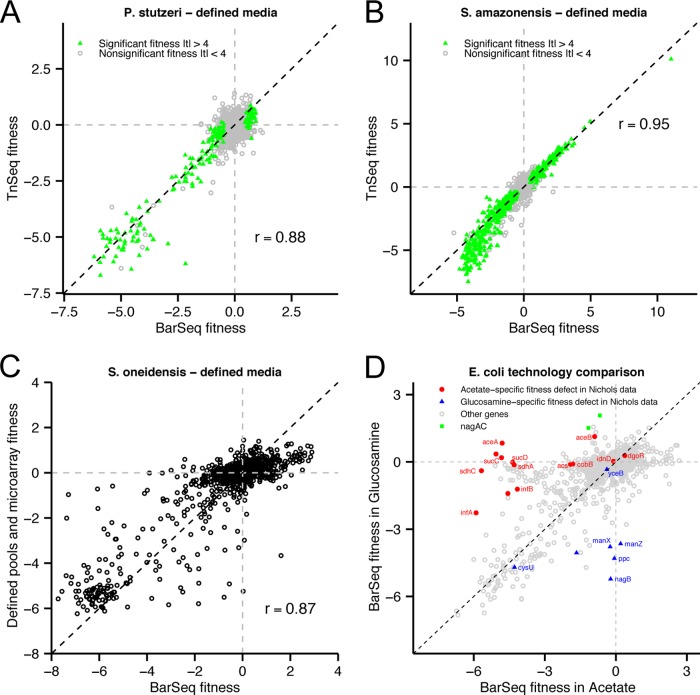
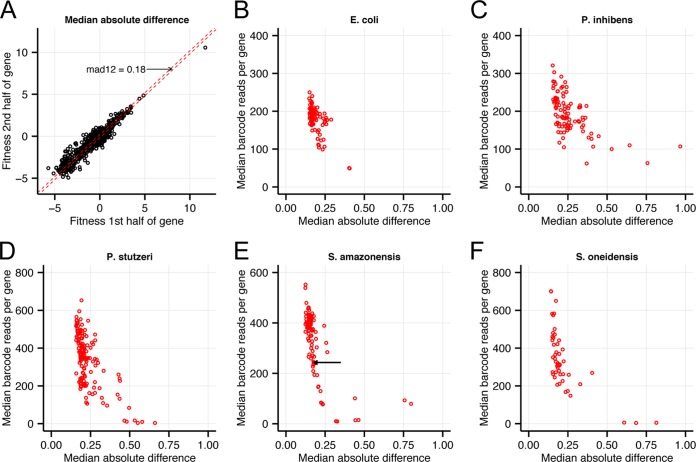
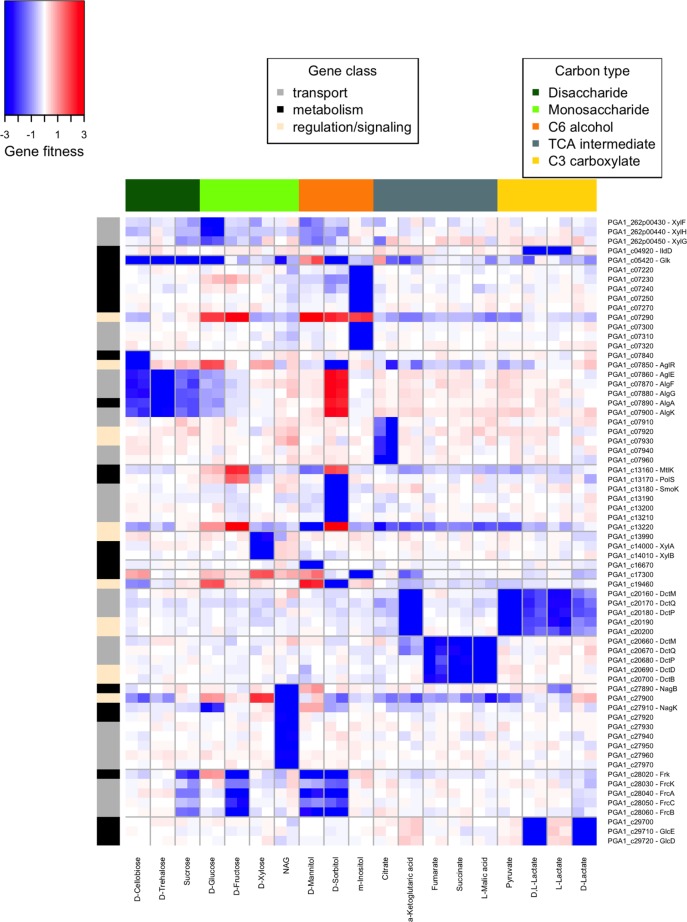
Transposon mutagenesis with next-generation sequencing (TnSeq) is a powerful approach to annotate gene function in bacteria, but existing protocols for TnSeq require laborious preparation of every sample before sequencing. Thus, the existing protocols are not amenable to the throughput necessary to identify phenotypes and functions for the majority of genes in diverse bacteria. Here, we present a method, random bar code transposon-site sequencing (RB-TnSeq), which increases the throughput of mutant fitness profiling by incorporating random DNA bar codes into Tn5 and mariner transposons and by using bar code sequencing (BarSeq) to assay mutant fitness. RB-TnSeq can be used with any transposon, and TnSeq is performed once per organism instead of once per sample. Each BarSeq assay requires only a simple PCR, and 48 to 96 samples can be sequenced on one lane of an Illumina HiSeq system. We demonstrate the reproducibility and biological significance of RB-TnSeq with Escherichia coli, Phaeobacter inhibens, Pseudomonas stutzeri, Shewanella amazonensis, and Shewanella oneidensis. To demonstrate the increased throughput of RB-TnSeq, we performed 387 successful genome-wide mutant fitness assays representing 130 different bacterium-carbon source combinations and identified 5,196 genes with significant phenotypes across the five bacteria. In P. inhibens, we used our mutant fitness data to identify genes important for the utilization of diverse carbon substrates, including a putative d-mannose isomerase that is required for mannitol catabolism. RB-TnSeq will enable the cost-effective functional annotation of diverse bacteria using mutant fitness profiling.
Importance: A large challenge in microbiology is the functional assessment of the millions of uncharacterized genes identified by genome sequencing. Transposon mutagenesis coupled to next-generation sequencing (TnSeq) is a powerful approach to assign phenotypes and functions to genes. However, the current strategies for TnSeq are too laborious to be applied to hundreds of experimental conditions across multiple bacteria. Here, we describe an approach, random bar code transposon-site sequencing (RB-TnSeq), which greatly simplifies the measurement of gene fitness by using bar code sequencing (BarSeq) to monitor the abundance of mutants. We performed 387 genome-wide fitness assays across five bacteria and identified phenotypes for over 5,000 genes. RB-TnSeq can be applied to diverse bacteria and is a powerful tool to annotate uncharacterized genes using phenotype data.
Copyright © 2015 Wetmore et al.
Figures
References
-
- Deutschbauer A, Price MN, Wetmore KM, Shao W, Baumohl JK, Xu Z, Nguyen M, Tamse R, Davis RW, Arkin AP. 2011. Evidence-based annotation of gene function in Shewanella oneidensis MR-1 using genome-wide fitness profiling across 121 conditions. PLoS Genet 7:e1002385. doi: 10.1371/journal.pgen.1002385. - DOI - PMC - PubMed
-
- Kuehl JV, Price MN, Ray J, Wetmore KM, Esquivel Z, Kazakov AE, Nguyen M, Kuehn R, Davis RW, Hazen TC, Arkin AP, Deutschbauer A. 2014. Functional genomics with a comprehensive library of transposon mutants for the sulfate-reducing bacterium Desulfovibrio alaskensis G20. mBio 5(3):e01041-14. doi: 10.1128/mBio.01041-14. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
